

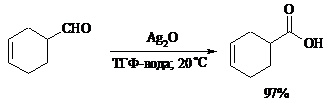
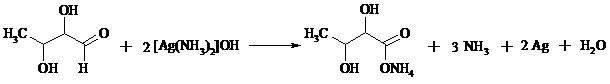
Окисление уксусного альдегида в промышленности осуществляют действием кислорода в присутствии солей кобальта или марганца в качестве катализатора:
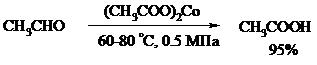
В химии углеводов для селективного окисления альдегидных групп применяется щелочной раствор иода. Так из D-глюкозы образуется D-глюконовая кислота:

Применяя разбавленную азотную кислоту, осуществляют окисление альдегидной группы, не затрагивая присутствующей в молекуле вторичной спиртовой группы, однако первичная спиртовая группа при этом окисляется в карбоксильную:

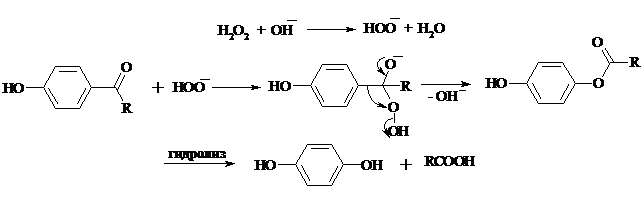
При обработке щелочным раствором пероксида водорода ароматических альдегидов (а также кетонов), содержащих в орто- или пара-положении NH2- или OH-группу, образуются соответственно аминофенолы или двухатомные фенолы (реакция Дакина):
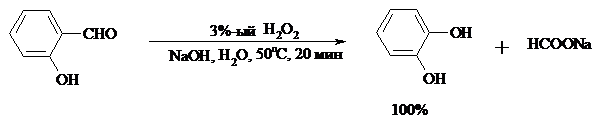
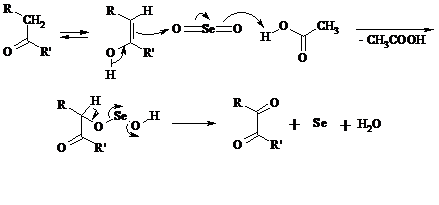
Под действием SeO2 альдегиды окисляются в a-дикарбонильные соединения:
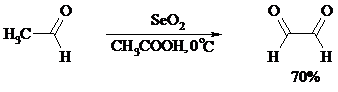
Интересно отметить, что при окислении диоксидом селена формальдегида образуется глиоксаль:
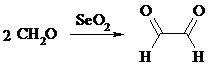
Альдегиды медленно окисляются кислородом воздуха при комнатной температуре до карбоновых кислот (аутоокисление альдегидов).
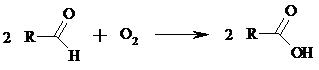
Эта реакция ускоряется при облучении или ионами Fe(II) и представляет собой цепной радикальный процесс:
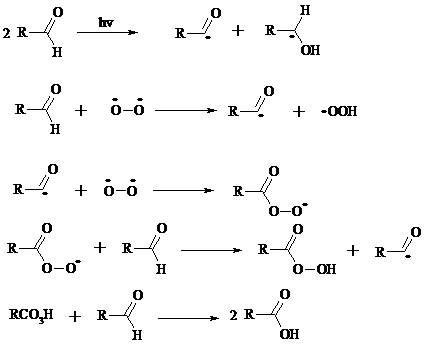
10. ОКИСЛЕНИЕ КЕТОНОВ
Окисление кетонов протекает в более жестких условиях, чем окисление альдегидов и сопровождается расщеплением углеродного скелета молекулы. Из несимметрично построенного кетона при окислении могут образоваться все четыре возможные карбоновые кислоты (правило Попова):

Если кетон в a-положении содержит третичный атом углерода, то в результате окисления образуется три карбоновые кислоты и новый кетон, который в зависимости от условий может или подвергнуться дальнейшему окислению, или остаться неизменным:

Кетоны, как правило, устойчивы к окислению в нейтральной среде и подвергаются расщеплению только в кислой и щелочной средах: щелочным раствором KMnO4, горячей HNO3, соединениями Cr(VI) в серной кислоте. В связи с этим считают, что окислению кетона предшествует его енолизация; образовавшийся енол подобно другим алкенам окисляется по кратной связи с расщеплением углеродного скелета. Поскольку несимметричные кетоны могут енолизоваться в обоих возможных направлениях, их окислительное расщепление также осуществляется по двум направлениям.
Промышленное значение имеет окисление циклогексанона, поскольку он легко доступен и при расщеплении дает только одну адипиновую кислоту. Реакцию проводят при повышенной температуре, применяя в качестве окислителя конц. HNO3, CrO3+H2SO4, щелочной раствор KMnO4:
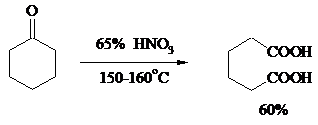
При окислении азотной кислотой алициклических кетонов температура реакции может быть снижена, а выход продукта повышен, если применять более разбавленную азотную кислоту и добавлять в реакционную смесь каталитические количества V2O5.
Циклические 1,3-дикетоны, которые существуют в основном в моноенольной форме, расщепляются периодатом натрия NaIO4 с потерей одного атома углерода:
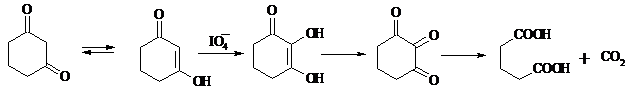
Метилкетоны легко превращаются в соответствующие карбоновые кислоты при обработке их щелочными растворами гипобромита или гипохлорита натрия, т. е. смесью галогена и щелочи (галоформная реакция). Таким образом из окиси мезитила можно, например, получить b, b-диметилакриловую кислоту:
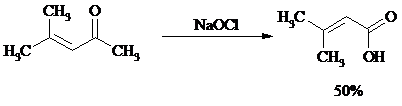
Метиларилкетоны окисляются с еще более высокими выходами. Реакция обычно проводится в водно-диоксановом растворе:
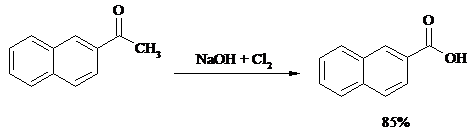
Механизм процесса можно представить следующим образом:
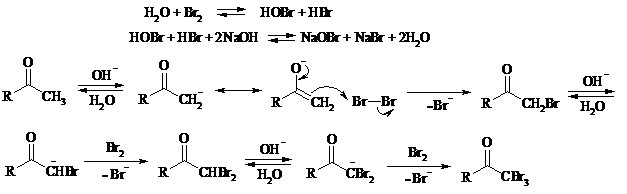
Далее под действием щелочи тригалогенметилкетон расщепляется по следующей схеме:
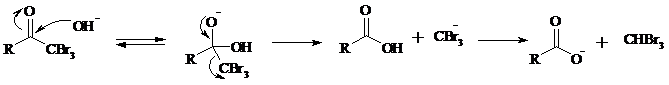
Под действием перкислот, а также кислоты Каро H2SO5 и H2S2O8 кетоны окисляются в сложные эфиры (реакция Байера-Виллигера). Реакция обычно проводится при комнатной температуре в полярных органических растворителях, устойчивых к действию надкислот (хлороформ, уксусная кислота, уксусный ангидрид):

Наилучшие результаты достигаются при расщеплении алифатических и циклических кетонов комплексом 90%-ного пероксида водорода и эфирата трехфтористого бора:
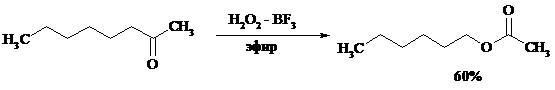
В начале под действием надкислоты, если она достаточно сильная, или каталитических количеств серной кислоты происходит протонизация карбонильного атома кислорода кетона. При этом карбонильный атом углерода приобретает избыточный положительный заряд. Затем к нему нуклеофильно присоединяется анион надкислоты. Последующий гетеролитический разрыв связи кислород-кислород, миграция одного из радикалов в виде аниона к положительно заряженному атому кислорода и отрыв протона приводят к образованию сложного эфира:
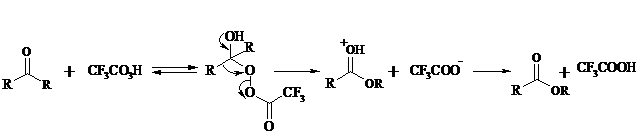
Несимметричные кетоны в общем случае образуют два изомерных сложных эфира, соотношение которых зависит от относительной способности групп к миграции к атому кислорода. По способности к миграции группы располагаются в следующем ряду H > C6H5 > (CH3)3C >> (CH3)2CH > RCH2 >> CH3. Если две группы сильно различаются по способности к миграции, реакция характеризуется высокой региоселективностью.
Особое значение имеет реакция Байера-Виллигера для циклических кетонов, которые при окислении дают циклические сложные эфиры – лактоны:
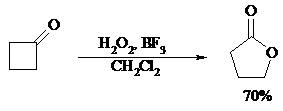
Метиленовая (метильная) группа в a-положении к карбонильной группе кетонов легко окисляется диоксидом селена:

Для реакции окисления кетонов, а также альдегидов диоксидом селена был предложен следующий механизм:
Вместо прямого окисления можно воспользоваться двухстадийным методом, приведенным ниже:
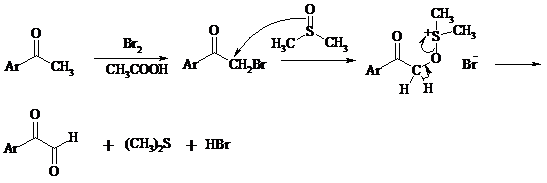
Алкоксисульфониевая соль, образующаяся при замещении галогена или тозилата, подвергается внутримолекулярному окислению-восстановлению с образованием кетоальдегида:
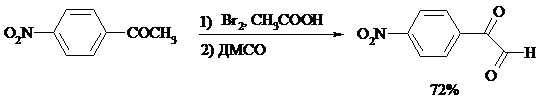
В то же время окисление α-бромкетонов диоксидом селена в спиртовой среде приводит к α-кетоэфирам:
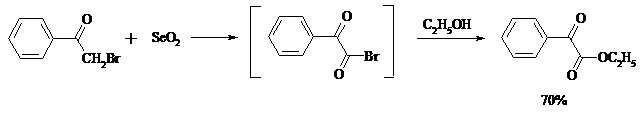
Под действием раствора полисульфида аммония (как правило, под давлением) арилалкилкетоны превращаются в ω-арилкарбоновые кислоты с сохранением общего числа атомов углерода (реакция Вильгеродта):
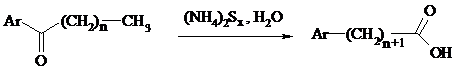
В результате карбонильная группа кетона восстанавливается до метиленовой, а метильная окисляется до карбоксильной. Фактически сначала получают тиоамид (или амид) кислоты, который затем омыляют. С ростом числа атомов углерода в цепи (n) выходы карбоновых кислот падают.
Улучшенный вариант реакции, не требующий применения давления, заключается в применении вместо полисульфида серы и вторичного амина (обычно морфолина) (модификация Киндлера):
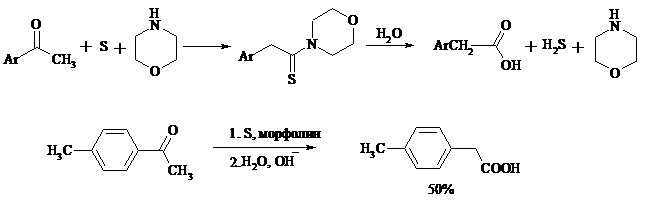
При обработке циклогексанонов серой или селеном протекает дегидрирование и образуются фенолы. По-видимому, реакция протекает через стадию предварительной енолизации исходных кетонов:
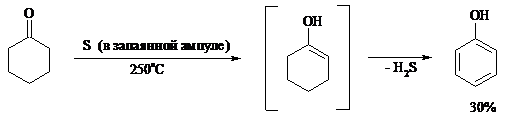
11. ОКИСЛЕНИЕ КАРБОНОВЫХ КИСЛОТ
При действии пероксида водорода в присутствии кислотного катализатора на карбоновые кислоты образуются перкислоты (надкислоты):
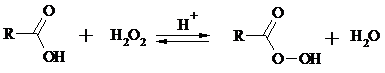
Наиболее распространенным катализатором в случае алифатических карбоновых кислот является концентрированная H2SO4. Реакция обратима, и равновесие можно сместить вправо, удаляя воду или применяя избыток реагента. Для субстратов с ароматическими группами R наилучшим катализатором является метансульфокислота, которая используется и как растворитель.
Карбоновые кислоты подвергаются окислительному декарбоксилированию под действием тетраацетата свинца и в качестве продуктов в зависимости от условий получаются алканы, алкены или эфиры уксусной кислоты:

Для данной реакции предполагается следующий механизм:
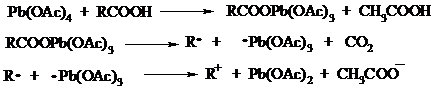
Алканы образуются за счет отрыва атома водорода от молекул растворителя радикалом R, а алкен и сложный эфир из карбокатиона соответственно за счет отщепления протона или захвата ацетат-иона. Введение в реакционную смесь галогенид-иона практически нацело подавляет оба процесса и приводит к образованию алкилгалогенидов.
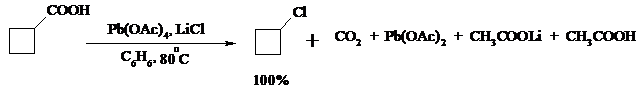
В присутствии иода и тетраацетата свинца карбоновые кислоты превращаются в соответствующие иодиды:
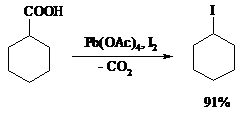
Из 1,2-дикарбоновых кислот под действием тетраацетата свинца образуются алкены:
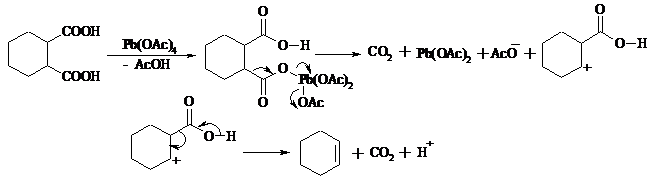
Другим примером окислительного декарбоксилирования может служить окисление серебряных солей карбоновых кислот бромом в CCl4 с образованием алкилбромидов (реакция Хунсдиккера – Бородина):
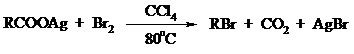
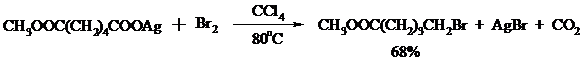
Для успешного проведения реакции требуется применять тщательно высушенные серебряные соли карбоновых кислот, и выход алкилбромида колеблется в широких пределах в зависимости от степени очистки и обезвоживания соли. Этого недостатка лишена модификация с использованием ртутных солей, причем ртутную соль не выделяют индивидуально, а смесь карбоновой кислоты, оксида ртути (II) и брома нагревают в индифферентном растворителе. Этот метод приводит, как правило, к более высоким и воспроизводимым выходам.
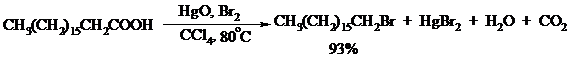
Для реакции Хунсдиккера – Бородина установлен цепной радикальный механизм. Образующийся в первой стадии ацилгипобромит подвергается гомолитическому расщеплению с образованием карбоксильного радикала и атома брома. Карбоксильный радикал теряет CO2 и превращается в алкильный радикал, который затем регенерирует цепь, отщепляя атом брома от ацилгипобромита.

a-Гидрокси - и a-кетокислоты не расщепляются под действием HIO4, но эта реакция идет с тетраацетатом свинца, H2O2 в щелочной среде и другими реагентами. Такие реакции представляют собой окислительное декарбоксилирование. Из a-гидроксикислот получаются альдегиды или кетоны, а из a-кетокислот – карбоновые кислоты:
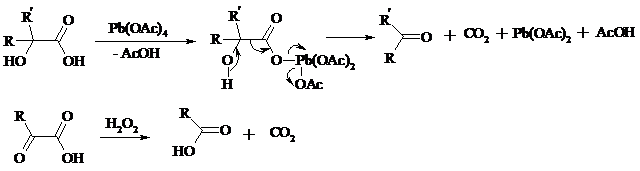
В то же время при использовании пероксида водорода в присутствии Fe(III) удается окислить гидроксильную группу в a-гидроксикислотах в карбонильную с сохранением карбоксильной:
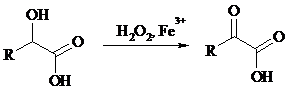
Окислить алифатические карбоновые кислоты в α-гидроксикислоты удается в том случае, если кислота содержит третичный атом углерода в α-положении к карбоксильной группе. В качестве окислителя применяют щелочной раствор KMnO4.
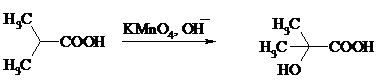
12. ОКИСЛЕНИЕ ПРОСТЫХ ЭФИРОВ
Простые эфиры проявляют повышенную склонность к аутоокислению в присутствии кислорода с образованием гидропероксидов. Процесс протекает по цепному радикальному механизму. Эффективным катализатором аутоокисления может служить любой источник свободных радикалов. Аутоокисление простых эфиров представляет большую потенциальную опасность при работе с эфирами в качестве растворителей, поскольку гидропероксиды, накапливающиеся в остатке при перегонке, могут детонировать при слабом перегреве.
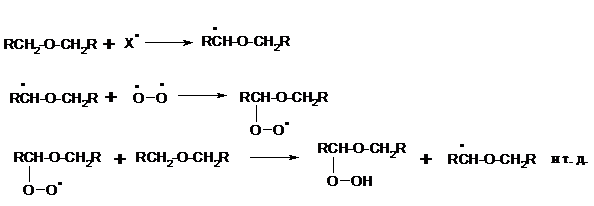
Поэтому гидропероксиды должны быть тщательно удалены до перегонки с помощью восстановителей – солей Fe(II) или Sn(II).
Простые эфиры, имеющие по крайней мере одну первичную алкильную группу, окисляются в соответствующие сложные эфиры с высокими выходами под действием RuO4, а также при действии CrO3 в серной кислоте, перманганата бензилтриэтиламмония и др.:
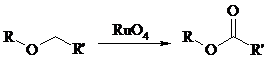
Простые эфиры енолов окисляются до сложных эфиров под действием хлорхромата пиридиния:
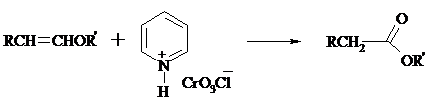
Под действием 1-хлорбензотриазола простые эфиры подвергаются окислительному расщеплению в альдегиды:
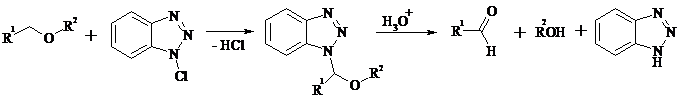
13. ОКИСЛЕНИЕ ЭПОКСИДОВ
HIO4 расщепляет эпоксиды в альдегиды и кетоны:
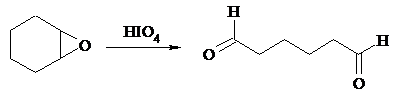
При окислении эпоксидов диметилсульфоксидом образуются a-гидроксикетоны или a-гидроксиальдегиды:
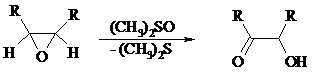
Сильные окислители превращают эпоксиды в кетоны и (или) карбоновые кислоты:
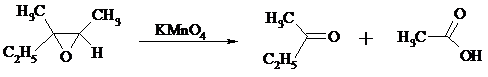
14. ОКИСЛЕНИЕ СЕРОСОДЕРЖАЩИХ СОЕДИНЕНИЙ
14.1. ОКИСЛЕНИЕ ТИОЛОВ
Под действием I2, Br2, H2O2, Pb(OAc)4, MnO2 тиолы окисляются до дисульфидов:
2RSH + I2 ® R-S-S-R
Перкислоты окисляют тиолы до сульфиновых кислот:
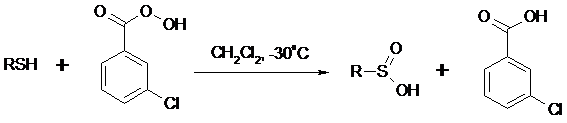
Сильные окислители – HNO3 и KMnO4 – окисляют тиолы до сульфоновых кислот:
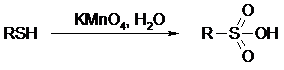
14.2. ОКИСЛЕНИЕ СУЛЬФИДОВ
Под действием метапериодата натрия NaIO4, м-хлорнадбензойной кислоты, трет-бутилгипохлорита сульфиды окисляются в сульфоксиды. Наиболее часто применяется 0.5 М водный раствор NaIO4. Этот реагент селективно окисляет сульфиды до сульфоксидов практически без примеси сульфонов, если окисление проводить при 0°С в бинарной системе вода – органический растворитель (метанол, диоксан, ацетонитрил):
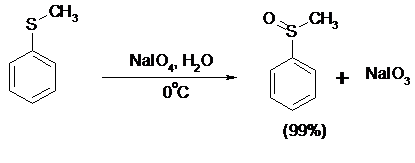
Механизм окисления аналогичен механизму расщепления 1,2-гликолей и включает циклический интермедиат:
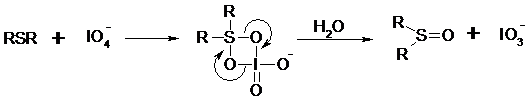
Превращение сульфидов в сульфоксиды под действием трет-бутилгипохлорита можно проиллюстрировать следующим примером:
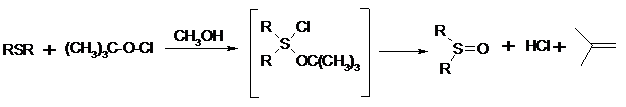
Окисление сульфидов до сульфонов осуществляется под действием более сильных окислителей (KMnO4, HNO3) или в более жестких условиях при 90-100°С с помощью избытка пероксида водорода или трет-бутилгидропероксида в уксусной кислоте:

Диоксираны селективно окисляют сульфиды до соответствующих сульфоксидов (при эквимольном соотношении реагентов) или до сульфонов (при избытке диоксирана).
15. ОКИСЛЕНИЕ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ
15.1. ОКИСЛЕНИЕ АМИНОВ
Все амины сравнительно легко окисляются из-за своей основной природы. Легче всего окисляются до N-оксидов третичные амины. В качестве окислителей используют 30%-ный раствор H2O2 в воде, перкислоты в апротонной среде, диоксираны:
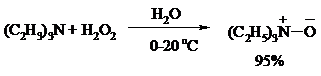
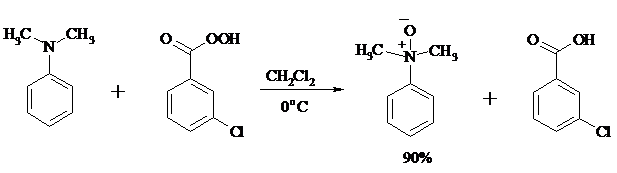
При окислении вторичных аминов вслед за образованием N-окиси происходит миграция протона с образованием N, N-диалкилгидроксиламина:
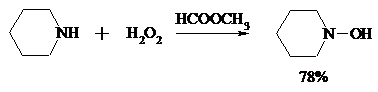
Первичные амины окисляются намного сложнее, поскольку образующееся производное гидроксиламина может далее окисляться до нитрозосоединений:

При наличии атома водорода при a-углеродном атоме нитрозосоединение изомеризуется в оксим:
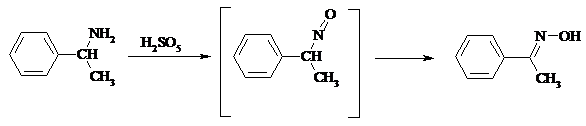
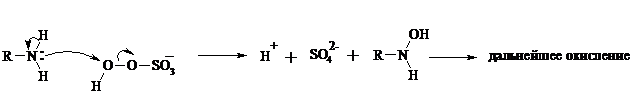
В более жестких условиях первичные амины окисляются до нитросоединений:

Окисление первичных аминов диметилдиоксираном протекает быстро (от нескольких минут до нескольких часов) в мягких условиях и приводит к соответствующим нитросоединениям с высоким выходом:
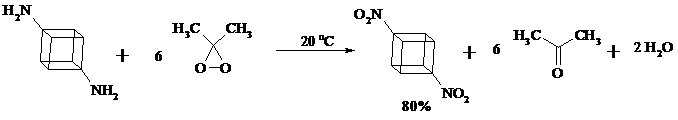
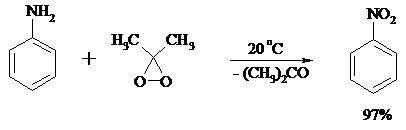
Первичные амины, в которых аминогруппа соединена с третичным атомом углерода, с высоким выходом окисляются в нитросоединения перманганатом калия. Первичные амины, содержащие первичные, вторичные или третичные алкильные радикалы окисляются до нитросоединений сухим озоном, а также различными перкислотами.
Первичные, вторичные и третичные алифатические амины расщепляются, давая альдегиды, кетоны или карбоновые кислоты, под действием бромной воды, N-бромсукцинимида, нейтрального раствора KMnO4, нитробензола (для бензиламинов), PdCl2, AuCl3, водного раствора NaOCl в условиях межфазного катализа:
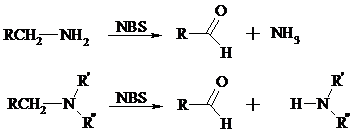
Первичные алифатические амины окисляются в альдегиды или кетоны при взаимодействии с Ag(II), полученным in situ обработкой нитрата серебра персульфатом натрия. Амин сначала дегидрируется до имина, который далее подвергается гидролизу:
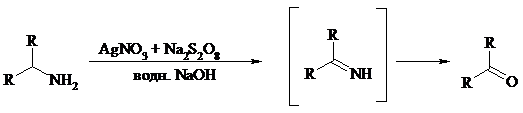
Соли бензиламинов дают бензальдегиды или арилкетоны при нагревании в ДМСО:
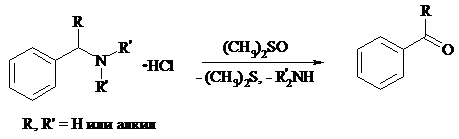
Дегидрирование первичной аминогруппы, соединенной с первичным атомом углерода, приводит к образованию нитрилов. Реакция осуществлена под действием ряда реагентов: IF5, Pb(OAc)4, CuCl – O2 – пиридин, NBS – (С2H5)3N и др.
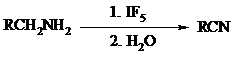
Вторичные амины в этих условиях, а также под действием палладиевой черни часто дегидрируются до иминов. Далее имин взаимодействует с молекулой исходного или иного амина, давая аминаль, который теряет аммиак или RNH2, и в результате получается первичный или третичный амин.
Ароматические первичные амины под действием MnO2, Pb(OAc)4, Ba(MnO4)2, O2 в присутствии основания окисляются до азосоединений:
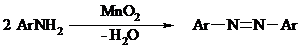
15.2. ОКИСЛЕНИЕ ГИДРАЗИНОВ, ГИДРАЗОНОВ, ОКСИМОВ,
ГИДРОКСИЛАМИНОВ И АЗОБЕНЗОЛОВ
N, N’-Диарилгидразины (гидразосоединения) окисляются в азосоединения при действии ряда окислителей, включая NaOBr, HgO, K3[Fe(CN)6] в условиях межфазного катализа, MnO2 (этот реагент приводит к цис-азобензолам), CuCl2, а также кислород воздуха в присутствии NaOH. Реакция применима также и к N, N’-диалкил - и N, N’-диацилгидразинам.
![]()
Гидразины (как алкильные, так и арильные), монозамещенные только с одной стороны, также дают азосоединения, однако последние неустойчивы и разлагаются на азот и углеводород:
![]()
При окислении гидразонов HgO, Ag2O образуются диазосоединения:
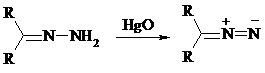
Гидразоны ароматических альдегидов взаимодействуют с HgO в таких растворителях, как диглим или этанол, давая нитрилы:
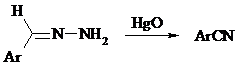
Окисление дигидразонов 1,2-дикарбонильных соединений является одним из лучших современных методов синтеза замещенных ацетиленов. В качестве окислителей используют HgO, Pb(OAc)4, O2 в присутствии СuCl и др.:
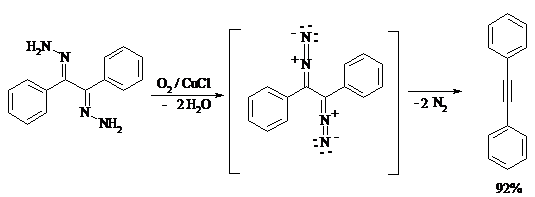
Ароматические гидроксиламины легко окисляются в нитрозосоединения чаще всего под действием Cr(VI) в кислой среде:
![]()
Окисление оксимов кетонов с помощью трифторперуксусной кислоты в ацетонитриле приводит к нитросоединениям. Первоначально при окислении оксимов образуется аци-форма нитросоединения, которая в кислой среде изомеризуется в нитросоединение:
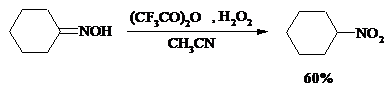
При окислении кетоксимов диметилдиоксираном также с высоким выходом образуются соответствующие кетоны:
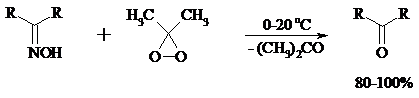
При действии N-бромсукцинимида на оксимы образуются геминальные бромнитрозосоединения (реакция Иффланда), которые далее могут быть окислены в бромнитросоединения под действием водного раствора гипохлорита, озона, HNO3 и др.:
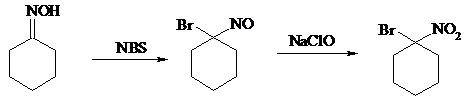
Продукты аналогичного строения могут быть получены в одну стадию при галогенировании кетоксимов бромом или хлором в щелочной среде:
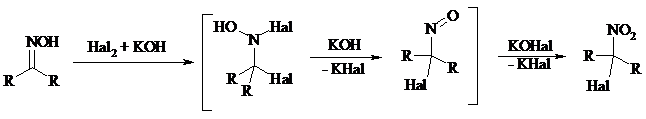
Азосоединения окисляются в азоксипроизводные перкислотами или гидропероксидами в присутствии комплексов молибдена:
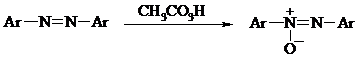
16. СИНТЕЗ НЕКОТОРЫХ ОКИСЛИТЕЛЕЙ
16.1. ДИХРОМАТ ПИРИДИНИЯ
2CrO3 + 2C5H5N + H2O → (C5H5NH)2Cr2O7
К 200 г (2.00 моль) оксида хрома (VI) в 200 мл воды прикапывают при перемешивании и охлаждении льдом 158 г (2.00 моль, 161 мл) пиридина. После этого прибавляют 800 мл ацетона и смесь охлаждают до -20ºС. Через 3 ч выпавшие оранжевые кристаллы отфильтровывают, промывают их ацетоном и сушат при комнатной температуре под вакуумом, получая 312 г (82%) продукта с т. пл. 149ºС.
Дихромат пиридиния – удобный в обращении нейтральный окислитель, использование которого позволяет превращать спирты в альдегиды и карбоновые кислоты.
16.2. ХЛОРХРОМАТ ПИРИДИНИЯ
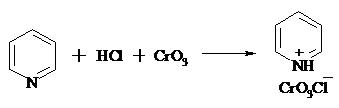
25 г (0.25 моль) оксида хрома (VI) при интенсивном перемешивании вносят в 46 мл 6 М соляной кислоты. Через 3 мин раствор охлаждают до 0ºС и в течение 10 мин при перемешивании прикапывают 19.7 г (0.25 моль, 20 мл) пиридина. Хлорхромат пиридиния, выпавший в виде оранжевых игл, быстро отфильтровывают на стеклянном фильтре и сушат 1 ч под вакуумом над оксидом фосфора, получая 45.2 г (84%) продукта. Реагент необходимо хранить в темноте без доступа влаги.
Хлорхромат пиридиния – хороший реагент для окисления первичных и вторичных спиртов до альдегидов и кетонов соответственно, обладает слабокислыми свойствами.
16.3. ХЛОРИСТЫЙ ХРОМИЛ
CrO3 + 2 HCl = CrO2Cl2 + H2O
К раствору 50 г (0.5 моль) CrO3 и 170 мл конц. HCl при охлаждении льдом прибавляют порциями по 20 мл 100 мл конц. H2SO4. Смесь жидкостей переливают в делительную воронку; через 20 мин сливают нижний слой CrO2Cl2 в круглодонную колбу со шлифом. Через жидкость в течение нескольких минут пропускают сухой воздух и затем перегоняют неочищенный CrO2Cl2 при атмосферном давлении в хорошо высушенном приборе на шлифах.
Хлористый хромил (диоксид-дихлорид хрома(VI)) – темно-красная жидкость, сильно дымящая во влажном воздухе. Хранят ее в запаянных стеклянных сосудах в темноте; т. пл. -96.5ºС, т. кип. 117ºС, d; растворим в CCl4, CHCl3, C6H6, POCl3.
16.4. ТЕТРААЦЕТАТ СВИНЦА
Pb3O4 + 8 CH3COOH = Pb(CH3COO)4 + 2 Pb(CH3COO)2
В двухлитровой трехгорлой колбе с мешалкой и термометром нагревают до 40ºС смесь 850 мл ледяной уксусной кислоты и 170 мл уксусного ангидрида, при сильном перемешивании вносят 343 г (0.5 моль) свинцового сурика Pb3O4. Температура при этом не должна подниматься выше 65ºС. Затем продолжают перемешивать при 60-65ºС до образования прозрачного раствора. При охлаждении выкристаллизовывается тетраацетат свинца. Его отфильтровывают, перекристаллизовывают из ледяной уксусной кислоты, сушат в вакуум-эксикаторе. Выход ~ 160 г.
Pb(CH3COO)4 легко гидролизуется с образованием диоксида свинца и уксусной кислоты, поэтому при кристаллизации и фильтровании его надо защищать от влаги воздуха.
16.5. ДИОКСИД СЕЛЕНА
В фарфоровой чашке нагревают на песчаной бане 50 мл концентрированной азотной кислоты и осторожно, небольшими порциями, вводят 30 г (0.38 моль) селена. Каждую новую порцию прибавляют после того, как закончилась реакция с предыдущей. При перемешивании смесь упаривают досуха, охлаждают, измельчают в порошок.
Активирование SeO2. Неочищенный диоксид селена помещают в фарфоровую чашку, приливают концентрированную азотную кислоту до образования густой “кашицы”. Чашку накрывают перевернутой воронкой и нагревают на песчаной бане. Сначала испаряются летучие продукты, затем при 315ºС возгоняется на стенки воронки SeO2. Скорость возгонки следует отрегулировать так, чтобы SeO2 не улетучивался через носик воронки. Для возгонки 40 г SeO2 требуется ~ 2.5 ч. Диоксид селена гигроскопичен.
Внимание! Диоксид селена очень токсичен, может вызывать поражение кожи.
16.6. АКТИВНЫЙ ДИОКСИД МАРГАНЦА
2 KMnO4 + 3 MnSO4 + 2 H2O = 5 MnO2 + K2SO4 + 2 H2SO4
1 способ. В колбу, снабженную капельной воронкой и механической мешалкой, вносят 250 мл 5%-ного раствора MnSO4 и при 50-60ºС приливают 200 мл 5%-ного раствора KMnO4 в течение 2 ч при перемешивании. Смесь нагревают при 60ºС еще 2 ч, после чего оставляют на сутки при 30-35ºС. Выпавший осадок отфильтровывают, промывают водой до удаления SO42- и сушат 8-10 ч при 110-120ºС. Выход 17 г (почти 100%).
2 способ. В стакане готовят суспензию 15 г тонко измельченного MnSO4 в 14 мл воды и при энергичном перемешивании медленно прибавляют 67.5 г 93%-ной H2SO4. Реакционная смесь разогревается. Температуру снижают до 50ºС и в течение 2 мин вносят небольшими порциями 15 г растертого KMnO4, следя, чтобы температура смеси не превышала 75ºС. Через 10 мин реакционную смесь выливают при перемешивании в сосуд с 2.5 л воды. Осадок диоксида марганца промывают водой сначала декантацией, затем на воронке до полного удаления SO42- и сушат 6 ч, постепенно повышая температуру до 180ºС. Выход около 28 г (почти 100%).
16.7. ФЕНИЛИОДОЗОДИАЦЕТАТ
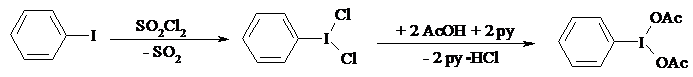
Иодбензол 20.4 г (0.1 моль, 11.2 мл) смешивают с 50 мл 98%-ной уксусной кислоты, при комнатной температуре приливают 20.25 г (0.15 моль, 12.2 мл) хлористого сульфурила. Смесь оставляют при комнатной температуре на 1 ч, выделившийся осадок фенилиодозохлорида фильтруют, на фильтре промывают петролейным эфиром. Выход 92%, т. пл. 117-119ºС.
При перемешивании растворяют 27.5 г (0.1 моль) фенилиодозохлорида в 65 мл пиридина, и раствор охлаждают до 0-5°С. Сохраняя эту температуру, постепенно из капельной воронки приливают 125 мл уксусной кислоты, потом 200 мл воды. Реакционную смесь перемешивают при 5-10ºС еще полчаса, затем фильтруют образовавшийся фенилиодозодиацетат, на фильтре промывают петролейным эфиром. Выход 24 г (75%). Продукт переосаждают из хлороформа петролейным эфиром, т. пл. 157-158°С.
16.8. ПЕРМАНГАНАТ КАЛИЯ НА ОКСИДЕ АЛЮМИНИЯ
В 5 мл воды растворяют 2.2 г (14.0 ммоль) тщательно растертого KMnO4 и добавляют при перемешивании 8.8 г оксида алюминия (активность по Брокману I). Смесь сушат на воздухе при комнатной температуре в течение суток.
16.9. ОКСИД СЕРЕБРА (I)
2 AgNO3 + 2 NaOH = Ag2O + 2 NaNO3 + H2O
Насыщенный раствор 10 г нитрата серебра обрабатывают в конической колбе раствором 2.5 г гидроксида натрия в 100 мл воды. Полученный темно-коричневый осадок тщательно промывают водой методом декантации, отжимают на воронке Бюхнера и сушат на воздухе при 60-80ºС. Выход количественный.
Выше 100ºС, а также на свету реактив разлагается на Ag и O2.
16.10. «МАЛИНОВЫЙ БЕНЗОЛ»
Тонко растертый KMnO4 суспендируют при комнатной температуре в бензоле и прибавляют эквимолярное количество дициклогексано-18-крауна-6. Раствор, приготовленный таким образом, можно довести упариванием до концентрации 0.06 моль/л.
16.11. 1-ХЛОРБЕНЗОТРИАЗОЛ
К раствору 95.84 г (0.80 моль) бензотриазола в 380 мл 50%-ной уксусной кислоте добавляют по каплям при перемешивании 1280 мл (0.96 моль) 5%-ного водного раствора NaOCl. После завершения добавления раствор перемешивают еще 2 ч, выпавший осадок отфильтровывают, промывают его водой до нейтральной среды и сушат в вакуум-эксикаторе. Выход 119 г (97%), т. пл. 103-105ºС.
17. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
17.1. n-БЕНЗОХИНОН
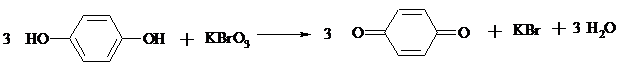
Реактивы | Посуда и приборы | ||
гидрохинон | 5 г | плоскодонная колба на 100 мл | |
5%-ный раствор H2SO4 | 2.5 мл | термометр | |
бромат калия | 2.75 г | колба Бунзена | |
воронка Бюхнера |
Внимание! n-Бензохинон токсичен, вызывает раздражение слизистых оболочек!
В плоскодонной колбе емкостью 100 мл растворяют 5 г (45.5 ммоль) гидрохинона в 50 мл воды, нагретой до 50ºС. Раствор охлаждают до 20°С и медленно при перемешивании по каплям прибавляют 2.5 мл 5%-ного раствора серной кислоты (катализатор). Затем в реакционную смесь вносят осторожно 2.75 г (16.5 ммоль) бромата калия, помещают термометр и нагревают на водяной бане до 60ºС. Начинается реакция окисления с образованием в качестве промежуточного продукта зеленовато-черного осадка хингидрона. Смесь выдерживают при 60ºС в течение 20 мин. Реакция окисления считается законченной, как только черный цвет реакционной массы изменится до ярко-желтого цвета n-бензохинона. В случае, если цвет не изменится полностью, следует добавить еще около 0.25 г бромата калия. Реакционную массу нагревают до полного растворения бензохинона (80ºС), затем охлаждают до 0ºС. Выпавший n-бензохинон отфильтровывают на воронке Бюхнера, промывают на фильтре небольшим количеством холодной воды и сушат на воздухе между листами фильтровальной бумаги.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
