

· изменения активности белков – трансмиттеров, аденилатциклазы, вторичных месенджеров, фосфодиэстераз, протеиназ и др. То есть, путем изменения активности участников пострецептороной передачи сигнала (рис. 12) клетка может ослабить или усилить эффект реагирования на него. Например, синтезируемые повреждения клеткой эйкозаноиды (см. выше) могут выполнять защитную функцию: связываясь с белком-трансмитером G (его ингибирующей субъединицей Gi) они препятствуют активации аденилциклазы (рис. 12). Таким образом, достигается «защита» клетки от целого ряда БАВ (катехоламинов, тиреоидных гормонов и др.);
· регуляции активности внутриклеточных метаболических процессов с помощью положительной или отрицательной связей. Интенсивность многих данных процессов контролируется количеством продукта ферментной реакции. Например, при гипоксии наблюдается снижение концентрации АТФ и увеличение продуктов ее распада – АДФ, АМФ. Данные продукты повышают активность одного из ключевых ферментов гликолиза – фосфофруктокиназу (ФФК) и клетки получают дополнительное количество энергии анаэробным путем окисления глюкозы. Количество АТФ увеличивается, а концентрация АДФ, АМФ – снижается и по механизму обратной отрицательной связи (при уменьшении гипоксии) активность ФФК падает, безкислородный метаболический путь синтеза АТФ угнетается.
Ликвидация дефектов генетического аппарата. Повреждение клетки нередко сопровождается изменением структуры ДНК, что ведет к нарушению реализации различных ее генетических программ.
В устранении дефектов генома и исправлении механизмов его реализации участвует ряд ферментов:
· эндонуклеазы (репарационная эндонуклеаза II) – обеспечивают обнаружение и удаление поврежденного участка ДНК;
· ДНК-полимеразы (ДНК-полимераза β) – осуществляют ресинтез нуклеиновой кислоты взамен удаленного участка;
· лигазы (ДНК-лигаза) – встраивают вновь синтезированный фрагмент ДНК на место удаленного.
Сравнительно редко встречаются генетические дефекты и в самой репарационной системе. Например, наследственная пигментная ксеродермия. Кожа данных больных очень чувствительна к солнечному свету, так как УФ-облучение провоцирует образование ферментов тимина (аминокислоты, входящей в одно из 4-х нуклеотидов ДНК). По мнению некоторых ученых и синдром преждевременного старения (прогерия) так же связан с ослаблением деятельности систем репарации ДНК.
Активация синтеза белков теплового шока (БТШ, НSР). Впервые они были обнаружены в клетках дрозофил, подвергшихся воздействию высоких температур. Позже выяснилось, что БТШ являются обязательными участниками ответа на повреждение различных клеток – от бактериальных до человеческих. Их синтез может быть инициирован не только гипертермией, но и гипоксией, воспалением, инфекцией, этиловым спиртом и др.
Белки теплового шока синтезируются в небольшом количестве и в норме (установлено, например, их участие в регуляции клеточной пролиферации). Повреждение клетки, как правило, сопровождается образованием денатурированного белка, который повышает активность генов, контролирующих синтез БТШ, и концентрация белков значительно возрастает.
В настоящее время известно несколько групп БТШ, различных по молекулярному весу и функциям. Защитный механизм белков теплового шока объясняется следующим:
· взаимодействуя с рецепторами стероидных гормонов, БТШ (БТШ 90кДа) предотвращают включение стрессорных программ клетки до наступления стресса. В условиях же последнего – смягчают (уменьшают) избыточную стимуляцию данными гормонами. Блокада рецепторов стероидных гормонов снижает апоптический эффект последних, благодаря чему, некоторые клетки при стрессе (например, лимфоциты) увеличивают продолжительность жизни. Таким образом, БТШ является своеобразным связующим звеном сопряжения стресса на уровне целостного организма и стрессового ответа отдельных клеток. Отсюда их еще одно название – «стрессовые белки»;
· осуществляя поддержку нативной конформации белков и сопровождение их после синтеза в различные отсеки клетки они тем самым предохраняют белки от агрегации и денатурации (БТШ 70кДа);
· мигрируя в ядро и связываясь с хроматином и ядрышком, БТФ тем самым, предохраняют появление мутаций и обеспечивают условия для восстановления повреждений ДНК;
· взаимодействуя с микротрубочками и микрофиламентами, стрессорные белки стабилизируют цитоскелет, что увеличивает устойчивость клетки к механическому повреждению, денатурации и агрегации белков клетки (БТШ 70кДа);
· по мнению , БТШ 15-30кДа повышают устойчивость клеток к острой гипоксии и развитию некроза (инфаркта миокарда). Данный эффект лежит в основе феномена, обозначенного им как «адаптационной стабилизацией структур»;
· БТШ 8,5-12кДа (самая низкомолекулярная группа) рассматриваются как рецепторы для специфических протеаз, способных активировать ограниченный протеолиз. С помощью ограниченного протеолиза осуществляется каскадная активация некоторых систем – свертывающей и противосвертывающей, системы комплемента и др. Предполагается, что данная группа БТШ необходима для ликвидации денатурированного белка и запуска протеолитических сигнальных систем в поврежденной клетке. Повышенный их синтез, может активировать эндонуклеазы и приводить к фрагментации хроматина, протеолизу циклина, что останавливает размножение дефектных клеток. Посредством активации (через протеолиз) белка р53 (белок-супрессор, останавливающий клеточное деление при повреждении ДНК) нередко срабатывает программа апоптоза.
Следовательно, система БТШ способна оказывать неспецифическое цитопротекторное действие при повреждении клетки, предохранять ее от включения механизмов апоптоза. Однако, при определенных условиях, процесс запрограммированной клеточной смерти может быть ими и инициирован.
Снижение функциональной активности поврежденной клетки. Приспособительные механизмы данного характера направлены на снижение степени и масштаба повреждения клетки при воздействии патогенного агента. Выполнение клеткой той или иной функции осуществляется не всей совокупностью ее структурных единиц. Часть из них, даже при интенсивной работе не используется. В этом заключается один из механизмов надежности любого образования (органа, ткани, клетки). Это так называемое статическое обеспечение надежности. Существует и другой, динамический механизм ее обеспечения.
Он определяется законом «перемежающейся активности функционирующих структур». Сущность закона – состав активных элементов (органа, клетки) в каждый момент, в процессе текущей реакции меняется, общее же количество действующих элементов может оставаться неизменным. Изменение состава (но не количества) происходит благодаря попеременному включению или выключению из реакции рабочих элементов. Благодаря данному закону при воздействии патогенного агента структуры клетки повреждаются в различной степени. Это происходит потому, что включении или выключение из активного состояния рабочего элемента происходит постепенно, т. е. они находятся в различном функциональном состоянии (но достаточном для выполнения функции). В то же время хорошо известно, что значительному повреждению (альтерации) подвергаются наиболее активно функционирующие структуры (рис. 13).
![]()

![]()


![]()
воздействия
 патогена
патогена
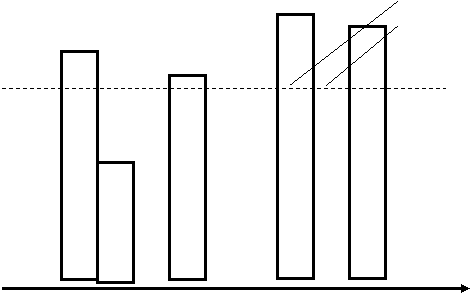 |
9 10 N
количество митохондрий
Рис. 13. Схема, иллюстрирующая (на примере митохондрий) значение закона «перемежающейся активности функционирующих структур» в патологии.
Десять митохондрий клетки, обеспечивающих достаточное количество АТФ, находятся в различном функциональном состоянии. В момент воздействия патогена самое большое повреждения будет наблюдаться в 1, 4 и 9 митохондриях находящиеся в 100% активности. Несколько меньше – 6, 7. Во 2, 5, 8, 10 митохондриях степень повреждения меньше, они будут, носит обратимый характер, и составят основу для восстановления энергообразования в клетке после прекращения (ослабления) действия патогенного агента
Поэтому, посредством уже знакомых нам механизмов (уменьшение количество сигнальных молекул, снижения числа и чувствительности мембранных и внутриклеточных рецепторов, репрессии некоторых клеточных программ и др.) клетка резко снижает активность, особенно выполнение своих специфических функций. Освободившиеся при этом ресурсы идут на поддержание адаптационных механизмов. Например, энергия, уходившая на выполнение специфической функции, используется для усиления компенсации изменений метаболизма, обусловленных повреждением (см. выше). То есть и перераспределение ресурсов значительно могут уменьшить степень повреждения клеток
Следовательно, врач всегда прав, когда больным назначает щадящий режим (постельный или амбулаторный), исключающий или резко снижающий физические и психологические нагрузки на больной организм.
Таким образом, снижение функциональной активности клетки, смягчает негативный эффект патогена и создает условия для более интенсивного и полного восстановления клеточных структур и их функций после его воздействия.
2. Защитно-приспособительные механизмы структурного характера.
Они формируются при длительном или периодическом воздействии патогенного фактора вызывающего легкие или средней тяжести повреждения. В этом случае, долговременная адаптация клеток обеспечивается за счет процессов регенерации, гипертрофии и гиперплазии. Регенерация (от лат. regeneratio возрождение, восстановление) – восстановление утраченной или поврежденной дифференцированной структуры. Различают клеточную и субклеточные формы регенерации. Первая проявляется размножением клеток, вторая – восстановление ее органелл (ядра, митохондрий, аппарат Гольджи и др.) вместо поврежденных или погибших.
Гипертрофия (от греч. hyper чрезмерно, увеличение + греч. trophe питание) – увеличения размера клетки и, как правило, внутриклеточных структур.
Гиперплазия (от греч. hyper чрезмерно + греч. plasis образование, формирование) – увеличение числа клеток, увеличение числа органелл. Нередко, в одной и той же клетке отмечаются признаки, и гиперплазии, и гипертрофии.
Основой для реализации гипертрофии и гиперплазии являются статическое и динамическое обеспечение надежности клетки (см. выше). Данные процессы могут обеспечить не только защиту (компенсацию) при утрате той или иной структуры, но и возможность повышенного функционирования клетки. Клетка может выполнять даже большую работу за счет пластического (структурного) усиления.
Для нормального развития процессов клеточной адаптации структурного плана необходима активация синтеза аминокислот, белка и др. Из них происходит в дальнейшем сборка клеточных органоидов и в целом образование новых клеток. И вот здесь имеет большое значение активация ряда генов (с-fos, c-jun, c-myc и др.) условно объединенных под названием «немедленные гены предранней реакции» (НГПР). Происходит это при любом повреждении клетки, и при достаточном количестве ростовых факторов (сигнальные молекулы). В ядре увеличивается активность орнитидиндекарбоксилазы и полиаминов (спермидин, спермин), последние регулируют синтез РНК и белка.
В то же время недостаток ростовых стимуляторов может запустить механизм апоптоза, а чрезмерная активация НГПР иногда приводить к малигнизации клетки. Например, ген c-myc является протоонкогеном, его повышенная экспрессия обнаруживается при злокачественной лимфоме Беркита (см. раздел «Злокачественный рост»).
Межклеточные (системные) механизмы адаптации.
В пределах органов, тканей и систем организма клетки не разобщены, они взаимодействуют между собой с помощью различных сигнальных молекул. Этим объясняется адекватный ответ нервной, эндокринной, сердечно-сосудистой и др. систем организма как целого на экзо - и эндогенные физиологические раздражители.
При заболеваниях межклеточные связи участвуют в реализации защитно-приспособительных реакций клетки. Однако, в отличии от внутриклеточных механизмов адаптации, они оказывают непрямой (опосредованный) эффект на процессы восстановления в поврежденной клетке. Наиболее часто это проявляется в виде ликвидации или ослаблении действия патогенного агента на клетку, и/или обеспечения ее всеми необходимыми питательными веществами (поврежденной клетки их нередко требуется больше, чем в норме). Причем, межклеточные механизмы осуществляются в основном клетками, не попавшими непосредственно под воздействие патогенного агента.
Выше мы отметили, что данные механизмы реализуются на:
· органно-тканевом уровне. Например, увеличение работы кардиомиоцитов за границами некроза при инфаркте миокарда, значительно снижает нагрузку на поврежденные клетки, уменьшает степень их дальнейшей альтерации и активирует процессы восстановления;
· внутрисистемном уровне. Например, при сердечной недостаточности, с целью предотвращения (уменьшения) снижения уровня артериального давления, происходит сужение периферических артериол. Данный механизм способствует поддержанию перфузии крови в органах и тканях и уменьшает воздействие на клетки такого патогенного фактора, как гипоксия;
· межсистемном уровне. Например, в ликвидации (снижении) негативного воздействия общего ацидоза на клетки принимают участие несколько систем организма. Система внешнего дыхания выводит избыток летучих кислот (СО2). Мочевыделительная система за счет ацидогенеза и аммониогенеза и др. механизмов увеличивает секрецию Н+ с мочой, повышает реабсорбцию из первичной мочи гидрокарбонатов. Активируется гидрокарбонатные и гидрофосфатные буферы костной ткани.
Мы закончили характеристику основных механизмов адаптации клетки. Благодаря им, клетка может вернуться к исходному уровню жизнедеятельности.
Однако, при их недостаточности, и, как было отмечено выше, вследствие своего несовершенства может наступить некроз (эндогенизация патологического процесса). Примеров, подтверждающих последнее положение, можно привести практически по каждому защитно-приспособительному механизму. Так, чрезмерная активация антиоксидантных систем клетки (направленных на снижение активности ПОЛ ее мембран) может значительно снизить резистентность к патогенной бактериальной флоре. Это происходит вследствие угнетения бактерицидного действия активными кислородными радикалами в фаголизосомах и их способности разрушать клеточные стенки бактерий. Анаэробный путь окисления глюкозы – это важный путь получения АТФ в условиях недостаточности кислорода. В то же время, длительная активация гликолиза нередко сопровождается развитием декомпенсированного внутриклеточного ацидоза со всеми, отсюда вытекающими последствиями для клетки. Или, как мы уже отмечали, повышенная активность немедленных генов предранней реакции (НГПР) может сопровождаться бластной трансфорацией клетки. Даже снижение функциональной активности поврежденной клетки, что очень положительно для нее, для состояния организма в целом имеет, в ряде случаев негативное значение. Например, уменьшение активности кардиомиоцитов снижает насосную функцию сердца и перфузия кровью органов и тканей падает.
В связи с этим, считаем необходимым еще раз напомнить – нередко защитная реакция организма имеет не только позитивное но и негативное значение. Врач должен уметь управлять развитием патологического процесса: каждую защитно-приспособительную реакцию он обязан рассматривать с данной позиции и по мере возможности исключать, уменьшать ее негативное воздействие.
8. НЕКРОЗ И АПОПТОЗ КЛЕТКИ
Различают два вида клеточной гибели: насильственная смерть от повреждения – некроз и запрограммированная клеточная смерть – апоптоз.
Некроз – это посмертные изменения клетки необратимого характера, заключающиеся в постепенном ферментативном разрушении и денатурации ее белков. Он развивается при чрезмерной альтерации клетки, не требует затрат энергии и не зависит от управляющих сигналов местного и центрального происхождения («анархических путь гибели»). Вследствие синтеза поврежденной клеткой БАВ (простогландины) и нарушения целостности ее мембран (выход различных ферментов), некроз представляет определенную угрозу окружающим структурам – это часто способствует развитию воспалительного процесса.
Насильственная гибель клетки обусловлена:
· лишением ее питания и кислорода;
· необратимыми изменениями структуры и функции с угнетением важнейших метаболических процессов различными патогенными агентами.
Некрозу предшествует глубокая, частично необратимая стадия повреждения клетки – некробиоз (рис. 1). Несмотря на многообразие этиологических факторов, провоцирующих в конечном счете развитие некробиоза и некроза, молекулярно-клеточные изменения, выявляемые при гибели клетки в большинстве случаев одинаковы (, , 1999). Согласно их мнению, важно различать гипоксический и свободно-радикальный некробиоз. Механизмы свободно-радикального повреждения клетки (см. выше) могут запускаться без первичной гипоксии, а иногда даже в условиях его избытка. Гипоксический некробиоз (см. раздел «Гипоксия») инициируется различными патогенными факторами, вызывающих продолжительную гипоксию. Оба вида некробиоза могут комбинироваться и взаимно дополнять друг друга. Исходом обоих видов некробиоза являются такие повреждения клетки, при которых она уже неспособна к самостоятельному энергообеспечению (т. необратимости, рис. 1) и подвергается некрозу.
Некоторые исследователи иногда рассматривают некробиоз, как процесс собственной гибели клетки. По , некробиоз – это процесс отмирания клеток. Некроз же, в большей степени характеристика морфологическая, наблюдающаяся после гибели клетки, а не механизм самой гибели.
Различают две основные разновидности некроза:
· коагуляционный (сухой) некроз. При нем в клетке развивается значительный ацидоз, идет коагуляция белков и отмечается повышенное накопление кальция с агрегацией элементов цитоскелета. Очень часто наблюдается при тяжелой гипоксии, например, в кардиомиоцитах при инфаркте миокарда. Данный некроз преимущественно развивается в тканях богатых белком и кальцием и характеризуется ранними и глубокими поражениями митохондрий;
· колликвационный некроз. Для него типично преобладание гидролитических процессов лизосомального аутолиза или гетеролизиса при участии фагоцитов. Очаг некроза размягчен, наблюдается накопление активных гидроксильных радикалов и эндогенное омыление клеток, что приводит к разрушению ее структур, например различных мембран.
Между коагуляционным и колликвационным некрозоми четких границ нет. Возможно, это объясняется тем, что механизмы их развития во многом общие. Ряд исследователей выделяют и так называемый казеозный (творожистый) некроз (при туберкулезе), пологая при этом, что он представляет собой комбинацию двух предыдущих типов.
Апоптоз – это программированная клеточная смерть (инициирующаяся под действием вне - или внутриклеточных факторов) в развитии которой активную роль принимают специальные и генетически запрограммированные внутриклеточные механизмы. Он, в отличие от некроза активный процесс, требующий определенных энергозатрат. Первоначально пытались разграничить понятия «программированная клеточная гибель» и «апоптоз»: к первому термину относили устранение клеток в эмбриогенезе, а ко второму – программированную смерть только зрелых дифференцированных клеток. В настоящее время выяснилось, что никакой целесообразности в этом нет (механизмы развития клеточной гибели одинаковы) и два понятия превратились в синонимы, хотя это объединение и не бесспорно.
Прежде чем приступить к изложению материала о роли апоптоза для жизнедеятельности клетки (и организма) в норме и патологии, мы рассмотрим механизм апоптоза. Их реализацию можно представить в виде поэтапного развития следующих стадий:
1 стадия – стадия инициации (индукции). В зависимости от происхождения сигнала, стимулирующего апоптоз, различают:
· внутриклеточные стимулы апоптоза. Среди них к наиболее известным относят – разные виды облучения, избыток Н+, оксид азота, свободные радикалы кислорода и липидов, гипертермия и др. Все они могут вызывать различные повреждения хромосом (разрывы ДНК, нарушения ее конформации др.) и внутриклеточных мембран (особенно митохондрий). То есть в данном случае поводом для апоптоза служит «неудовлетворительное состояние самой клетки» (, , 2003). Причем, повреждение структур клеток должно быть достаточно сильным, но не разрушительным. У клетки должны сохраниться энергетические и материальные ресурсы для активации генов апоптоза и его эффекторных механизмов. Внутриклеточный путь стимуляции программированной смерти клетки можно обозначить как «апоптоз изнутри»;
· трансмембранные стимулы апоптоза, т. е., в этом случае он активируется внешней «сигнализацией», которая передается через мембранные или (реже) внутриклеточные рецепторы. Клетка может быть вполне жизнеспособной, но, с позиции целостного организма или «ошибочной» стимуляции апоптоза, она должна погибнуть. Этот вариант апоптоза получил название «апоптоз по команде».
Трансмембранные стимулы подразделяются на:
· «отрицательные» сигналы. Для нормальной жизнедеятельности клетки, регуляции ее деления и размножения необходимо воздействие на нее через рецепторы различных БАВ: факторов роста, цитокинов, гормонов. Среди прочих эффектов, они подавляют механизмы клеточной гибели. И естественно, дефицит или отсутствие данных БАВ активирует механизмы программированной смерти клетки;
· «положительные» сигналы. Сигнальные молекулы, такие как ФНОα, глюкокортикоиды, некоторые антигены, адгезивные белки и др., после взаимодействия с клеточными рецепторами могут запускать программу апоптоза.
На клеточных мембранах находится группа рецепторов, в задачу которых передача сигнала к развитию апоптоза является основной, возможно даже единственной функцией. Это, например, белки группы DR (death receptos – «рецепторы смерти»): DR3, DR4, DR5. Наиболее хорошо изучен Fas-рецептор, появляющийся на поверхности клеток (гепатоцитах) спонтанно или под влиянием активации (зрелые лимфоциты). Fas-рецептор при взаимодействии с Fas-рецептором (лигандом) Т-киллера запускает программу смерти клетки мишени. Однако, взаимодействие Fas-рецептора с Fas-лигандом в областях, изолированных от иммунной системы, заканчивается гибелью самого Т-киллера (см. нижеигандом в областях, изолированных от иммунной системы, заканчивается гибелью самого Т-киллера ()ожно).
Следует помнить, что некоторые сигнальные молекулы апоптоза, в зависимости от ситуации могут наоборот, блокировать развитие программированной смерти клеток. Амбивалентность (двойственное проявление противоположных качеств) характерна для ФНО, ИЛ-2, интерферона γ и др.
На мембранах эритроцитов, тромбоцитов, лейкоцитов, а так же клеток легкого и кожи обнаружены особые антигены-маркеры. На них синтезируются физиологические аутоантитела, и они, выполняя роль опсонинов, способствуют фагоцитозу этих клеток, т. е. гибель клеток происходит путем аутофагоцитоза. Выяснилось, что антигены-маркеры появляются на поверхности «старых» (прошедших свой путь онтогенетического развития) и поврежденных клетках, молодые и неповрежденные клетки их не имеют. Данные антигены получили название «антигены-маркеры стареющих и поврежденных клеток» или «белок третьей полосы». Появление белка третьей полосы контролируется геномом клетки. Следовательно, аутофагоцитоз можно рассматривать, как вариант запрограммированной гибели клеток.
· Смешанные сигналы. Это сочетанное воздействие сигналов первой и второй группы. Например, апоптоз происходит с лимфоцитами, активированных митогоном (положительный сигнал), но не вступивших в контакт с АГ (отрицательный сигнал).
2 стадия – стадия программирования (контроля и интеграции механизмов апоптоза).
Для этой стадии характерно два, диаметрально противоположных процесса, наблюдающихся после инициации. Происходит либо:
· реализация пускового сигнала к апоптозу через активацию его программы (эффекторами являются каспазы и эндонуклеазы);
· блокируется эффект пускового сигнала апоптоза.
Различают два основных, но не исключающих друг друга, варианта исполнения стадии программирования (рис. 14):
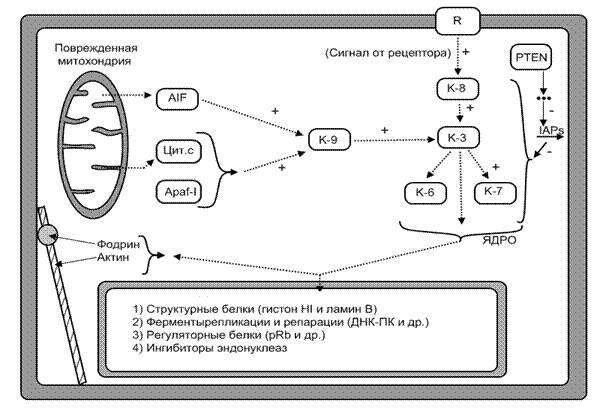
Рис. 14. Каспазный каскад и его мишени
R – мембранный рецептор; К – каспазы; AIF – митохондриальная протеаза; Цит. С – цитохром с; Apaf-1 – цитоплазматический белок; IAPs – ингибиторы каспаз
1. Прямая передача сигнала (прямой путь активации эффекторных механизмов апоптоза минуя геном клетки) реализуется через:
· адапторные белки. Например, так осуществляется запуск апоптоза Т-киллером. Он активирует каспазу-8 (адапторный белок). Аналогично может действовать и ФНО;
· цитохром С и протеазу ΑIF (митохондриальная протеаза). Они выходят из поврежденной митохондрии и активируют каспазу-9;
· гранзимы. Т-киллеры синтезируют белок перфорин, который образует каналы в плазмолемме клетки-мишени. Через эти каналы в клетку проникают протеолитические ферменты гранзимы, выделяемые все тем же Т-киллером и они запускают каскад каспазной сети.
2. Опосредованная передача сигнала. Она реализуется с помощью генома клетки путем:
· репрессии генов, контролирующих синтез белков-ингибиторов апоптоза (гены Bcl-2, Bcl-XL и др). Белки Bcl-2 в нормальных клетках входят в состав мембраны митохондрий и закрывают каналы по которым из этих органоидов выходят цитохром С и протеаза AIF;
· экспрессии, активации генов, контролирующих синтез белков-активаторов апоптоза (гены Bax, Bad, Bak, Rb, P53 и др.). Они, в свою очередь активируют каспазы (к-8, к-9).
На рис. 14 представлена примерная схема каспазного принципа активации каспаз. Видно, что откуда бы не запускался каскад, его узловым моментом является каспаза 3. Она активируется и каспазой 8 и 9. Всего в семействе каспаз – более 10 ферментов. Локализуются в цитоплазме клетки в неактивном состоянии (прокаспазы). Положение всех каспаз в данном каскаде до конца не выяснено, поэтому на схеме ряд из них отсутствует. Как только активируются каспазы 3,7,6 (возможно и их другие типы) наступает 3 стадия апоптоза.
3 стадия – стадия реализация программы (исполнительная, эффекторная). Непосредственными исполнителями («палачами» клетки) являются выше указанные каспазы и эндонуклеазы. Местом приложения их действия (протеолиза) служат (рис. 14):
· цитоплазматические белки – белки цитоскелета (фодрин и актин). Гидролизом фодрина объясняют изменение поверхности клетки – «гофрирование» плазмолеммы (появление на ней впячиваний и выступов);
· белки некоторых цитоплазматических регуляторных ферментов: фосфолипазы А2, протеинкиназы С и др.;
· ядерные белки. Протеолиз ядерных белков занимает основное место в развитии апоптоза. Разрушаются структурные белки, белки ферментов репликации и репарации (ДНК-протеинкиназы и др.), регуляторные белки (рRb и др.), белки-ингибиторов эндонуклеаз.
Иннактивация последней группы – белков ингибиторов эндонуклеаз приводит к активации эндонуклеаз, второму «орудию» апоптоза. В настоящее время эндонуклеазы и в частности, Са2+, Мg2+ - зависимая эндонуклеаза, рассматривается как центральный фермент программируемой смерти клетки. Она расщепляет ДНК не в случайных местах, а только в линкерных участках (соединительные участки между нуклеосомами). Поэтому хроматин не лизируется, а только фрагментируется, что определяет отличительную, структурную черту апоптоза.
Вследствие разрушения белка и хроматина в клетке формируются и от нее отпочковываются различные фрагменты – апоптозные тельца. В них находятся остатки цитоплазмы, органелл, хроматина и др.
4 стадия – стадия удаления апоптозных телец (фрагментов клетки). На поверхности апоптозных телец экспрессируются лиганды, они распознаются рецепторами фагоцитов. Процесс обнаружения, поглощения и метаболизирования фрагментов погибшей клетки происходит сравнительно быстро. Это способствует избежать попадания содержания погибшей клетки в окружающую среду и тем самым, как отмечено выше, воспалительный процесс не развивается. Клетка уходит из жизни «спокойно», не беспокоя «соседей» («тихий суицид»).
Программированная клеточная гибель имеет важное значение для многих физиологических процессов. С апоптозом связаны:
· поддержание нормальных процессов морфогенеза – запрограммированная смерть клеток в процессе эмбриогенеза (имплантации, органогенеза) и метаморфоза;
· поддержание клеточного гомеостаза (в том числе ликвидация клеток с генетическими нарушениями и инфицированных вирусами). Апоптозом объясняется физиологическая инволюция и уравновешивание митозов в зрелых тканях и органах. Например, гибель клеток в активно пролиферирующих и самообновляющихся популяциях – эпителиоцитов кишечника, зрелых лейкоцитов, эритроцитов. Гормонально-зависимая инволюция – гибель эндометрия в конце менструального цикла;
· селекция разновидностей клеток внутри популяции. Например, формирование антигенспецифической составляющей иммунной системы и управление реализацией ее эффекторных механизмов. С помощью апоптоза происходит выбраковка ненужных и опасных для организма клонов лимфоцитов (аутоагрессивных). Сравнительно недавно (Griffith T. S., 1997) показали значение программированной гибели клеток в защите «иммунологически привилегированных» зон (внутренние среды глаза и семенников). При прохождении гисто-гематических барьеров данных зон (что случается редко), эффекторные Т-лимфоциты гибнут (см. выше). Включение механизмов их смерти обеспечивается при взаимодействии Fas-лиганда барьерных клеток с Fas-рецепторами Т-лимфоцита, тем самым предотвращается развитие аутоагрессии.
Роль апоптоза в патологии и виды различных заболеваний связанных с нарушением апоптоза представлены в виде схемы (рис. 15) и таблицы 1.
Конечно, значение апоптоза в патологии меньше чем некроза (возможно, это связано с недостаточностью таких знаний). Однако, проблема его в патологии имеет и несколько иной характер: она оценивается по степени выраженности апоптоза - усиление или ослабление при тех или иных болезнях.
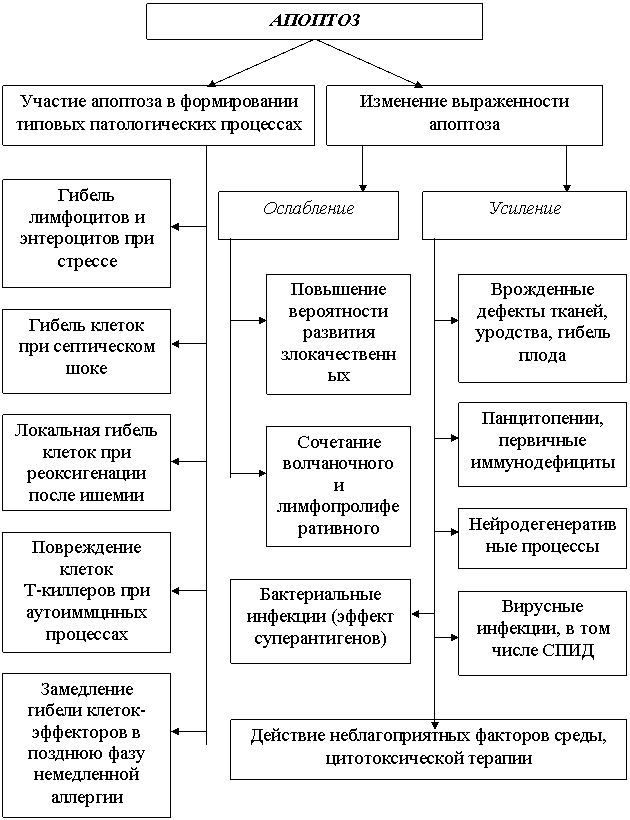
Рис. 15. Роль апоптоза в патологии
Таблица 1
Патология, связанная с нарушением апоптоза
Группа заболеваний | Заболевания | Комментарии |
Заболевания, связанные с ослаблением апоптоза | ||
Аутоиммунные процессы Злокачественные опухоли | Семейный аутоиммунный лимфопролиферативный синдром, связанный с дефицитом Fas Системная красная волчанка, ревматоидный артрит, синдром Бехчета Лимфома Беркита Лейкозы и солидные опухоли | Аналоги у мышей – носители мутации генов lpr, gld и трансфектанты по гену bcl-2. СКВ-подобный синдром в сочетании с накоплением клеток CD3+CD4-CD8-B220+ Ослабление апоптоза невыясненного генеза. Образование растворимого Fas-рецептора Транслокация и гиперэкспрессия генов bcl-2 и c-myc; ослабление апоптоза Мутация и экспрессия гена р53 приводят к ослаблению апоптоза, что часто коррелирует с прогрессированием и их устойчивостью к терапии |
Заболевания, связанные с усилением апоптоза | ||
Врожденные аномалии Болезни крови (цитопении) Инфекционные (бактериальные заболевания) Вирусные инфекции Дистрофические заболевания нервной системы Другие заболевания | Синдрома Дауна и др. Миелодисплазии, апластическая, Fе-, фолат, В12-дефицитные анемии, тромбоцитопения, нейтропения, болезнь Кастлемана Различные инфекционные процессы, сепсис Различные вирусные заболевания, в том числе СПИД Боковой амиотрофический склероз, болезнь Альцгеймера, спинальная мышечная атрофия Инфаркт миокарда Токсические гепатиты | Преобладание апоптоза при формировании локальных структур Усиление апоптоза клеток отдельных или всех ростков кроветворения в процессе развития Апоптоз клеток иммунной системы вызывают суперантигены, токсины; при сепсисе – накапливающийся в крови ФНОα Индукторами апоптоза при СПИДе служат вирусные факторы, в частности gр120, взаимодействующий с CD4 Усиление апоптоза нейронов и других клеток в определенных участках центральной нервной системы Преобладание апоптоза миоцитов в период «репефузии» миокарда Апоптоз гепатоцитов под действием ядов, в том числе этанола |
Усиление апоптоза характеризуется развитием патологии, затрагивающей в основном определенные типы клеток с уменьшением их численности (СПИД, тромбоцитопения, болезнь Альцгеймера и др.). Недостаточность механизмов программированной гибели клеток лежит в основе заболеваний, в развитии которых решающим является проявление активности клеток, в норме подлежащих ограничению или даже полной ликвидации (аутоиммунные процессы, доброкачественные и злокачественные опухоли). Данные изменения механизмов апоптоза, естественно нужно учитывать при разработке патогенетической терапии.
В последнее время, область фармакологии, посвященная разработке лекарственных препаратов по регулированию и коррекции механизмов программированной клеточной гибели, успешно формируется. Сейчас наши знания по механизмам апоптоза значительно расширились и появилась уверенность, что успехи фармакологии апоптоза получат широкое внедрение в практическую медицину.
9. ОСНОВЫ ЭТИОТРОПНОЙ И ПАТОГЕНЕТИЧЕСКОЙ ПРОФИЛАКТИКИ И ТЕРАПИИ ПАТОЛОГИИ КЛЕТКИ.
Этиотропная профилактика (первичная профилактика). Она включает в себя мероприятия и средства направленные на предотвращение (уменьшение) воздействия различных причинных факторов и условий способствующих повреждению клеток. Они используются до воздействия патогенного агента на клетку и включают в себя:
· немедикаментозные средства. Широкое распространение получили методы, направленные на повышение неспецифической резистентности клеток (и организма в целом) к большой группе патогенных агентов. Используя методы тренировки организма (по специально разработанным схемам) к умеренной гипоксии (метод нормобарической гипоксии), физическим нагрузкам, гипотермии (охлаждении) и др. можно значительно повысить устойчивость клеток. Помимо того, что тренировки усиливают резистентность клеток к воздействию тяжелой гипоксии, ионизирующей радиации, переохлаждению, инфекционным возбудителям и др. причинным агентам, они активируют и процессы восстановления (репарации).
Повышение устойчивости клеток организма в данном случае обеспечивается за счет активации защитно-приспособительных механизмов структурного характера (см. выше). То есть, клетка, еще до встречи с патогенным агентом, получает структурно-функциональное усиление, что позволяет в условиях взаимодействия с ним значительно уменьшить степень повреждения, либо вообще не иметь таковых.
Используются и специфические мероприятия, направленные на предотвращение или ослабление воздействия определенных патогенных агентов. Например, наличие спецодежды и экранирования источника радиоактивного излучения является обязательным средством защиты от ионизирующего поражения:
· химические средства (дезинфицирующие вещества, биологически активные добавки – БАД, лекарственные препараты). Они наиболее эффективны при прямой угрозе поражения клеток тем или иным агрессивным фактором. Например, для предупреждения повреждения биологической группой этиологических факторов используют различные дезинфицирующие вещества или назначают антибиотики после обширных травм, в послеоперационном периоде. БАД (витамины С, РР, адаптогены), лекарственные препараты (цистимин, мексамин и др.) защищают клетку от ионизирующего излучения (радиопротекторный эффект).
Этиотропная терапия. Включает в себя все мероприятия направленные на прекращение либо уменьшение интенсивности воздействия и/или длительности действия причинного фактора на клетку. Классическим примером данной терапии являются лекарственные средства (антибиотики, сульфаниламиды) применяемые против биологических факторов повреждения. Устранение недостатка или избытка того или иного ингредиента пищи (аминокислот, жиров, микроэлементов и др.) способствующего нарушению функции клеток, так же явления предметом этиотропной терапии.
Патогенетическая профилактика (вторичная профилактика). Она направлена на предупреждение (уменьшение) прогрессирования патологии клетки и неотделима от патогенетической терапии. Применяется с учетом знаний механизмов патогенеза конкретного вида повреждения клетки. Например, для уменьшения степени повреждения мембран клетки вводят блокаторы фосфолипаз, протеаз (делагил, никотинамид), мембранностабилизирующие препараты.
Патогенетическая терапия. Ее основными задачами являются:
1. Воздействие на различные патогенетические звенья повреждения клетки для ликвидации и/или уменьшения их негативного эффекта. Используют различные лекарственные средства, с целью:
· устранения или уменьшения степени нарушений энергообеспечения клетки (ингаляция О2, глюкозо-инсулино-калиевая смесь, карнитин, антигипоксанты и др.);
· стабилизации (защиты) плазмолеммы и ферментов клетки - антигипоксанты, антиоксиданты (токоферолы, маннитол и др.), антогонисты кальция, блокаторы фосфолипаз, липаз, протеаз, глюкокортикоиды и др.;
· коррекции водно-электролитного гомеостаза клетки – лекарственные препараты регулирующие трансмембранный перенос К+ и Nа+ (лидокаин, строфантин, К+содержащие средства и др.), антогонисты Са2+, осмотически активные и буферные растворы (бикарбонаты, фосфаты, маннитол и др.);
· оптимизации регуляторных влияний на клетку и интеграции внутриклеточных процессов в изменившихся условиях (в условиях патологии) применяют гормоны, нейромедиаторы и другие сигнальные молекулы, методы и схемы их назначения зависят от характера патологии клетки.
2. Активация саногенетических (защитно-приспособительных) механизмов, что способствует повышению резистентности клетки. Они обеспечивают компенсацию, восстановление и приспособление клетки в изменившихся условиях. Например, при рассмотрении вопросов этиотропной профилактики, мы отметили эффективность метода тренировок организма к различным воздействиям (гипоксии, гипотермии и др.). Этот же метод, применяемый после исчезновения проявлений острых повреждений клеток, значительно ускоряет процессы восстановления и репарации.
10. ЗАДАНИЕ ДЛЯ САМОКОНТРОЛЯ
Тестовые задания:
1. Выберите наиболее правильный ответ:
1) паранекроз – это гибель клетки в течение нескольких часов;
2) паранекроз – это начальные, обратимые стадии повреждения клетки;
3) паранекроз – это гибель клетки в течение нескольких суток;
4) паранекроз – это необратимые повреждения клетки, предшествующие ее некрозу.
2. К биохимическим критериям гибели клетки относят:
1) выход ферментов лизосом в цитоплазму клетки;
2) выход ферментов лизосом в межклеточное пространство;
3) полное прекращение синтеза АТФ;
4) выход ферментов цитозоля в межклеточное пространство.
3. Перечислите типы (виды) повреждения клеток:
1) митохондриальные;
2) интерфазные;
3) лизосомальные;
4) специфические;
5) неспецифические.
4. Повреждение мембраны клеток может быть результатом:
1) активации свободнорадикального окисления;
2) активации мембранносвязанных фосфолипаз;
3) снижение синтеза простогландинов;
4) адсорбция на мембране полиэлектролитов.
5. Эффекты чрезмерной активации СР и ПОЛ следующие:
1) иннактивация карбоксильных и сульфгидрильных групп белков плазмолеммы;
2) формирование «кластеров» в клеточной мембране;
3) снижение проницаемости мембраны для ионов;
4) ингибирование активности мембранных фосфолипаз.
6. Назовите антиоксидантные ферменты:
1) каталаза;
2) супероксикаталаза;
3) глутатион-пероксидаза;
4) пируваткиназа.
7. Основными последствиями соматических мутаций могут быть:
1) активация механизмов апоптоза;
2) болезнь Дауна;
3) наследственные энзимопатии;
4) изменение антигенной структуры клеток.
8. Разобщение процессов окисления и фосфорилирования в митохондриях вызывают:
1) простогландины;
2) паратгормон;
3) тироксин;
4) билирубин.
9. Назовите заболевания, относящиеся к болезням накопления (тезаурисмозам):
1) болезнь Помпе;
2) болезнь Крассе;
3) болезнь Крабе;
4) болезнь Таусики.
10. Патология гладкого эндоплазматического ретикулума характеризуется:
1) нарушением синтеза мембранных липидов;
2) нарушением синтеза мембранных белков;
3) нарушением модификации белков;
4) нарушением дезинтоксикационных процессов клетки.
11. Развитию гипергидротации клетки способствуют:
1) снижение синтеза АТФ;
2) повышение активности К+, Nа+-АТФазы;
3) увеличение содержания К+ в клетке;
4) уменьшение внеклеточного содержания Nа+.
12. «Болезни регуляции» – это результат:
1) нарушения рецепции сигналов;
2) патологии сигнализации;
3) патологии митохондрий;
4) нарушения работы К+/Nа+ АТФазы.
13. Нарушение пострецепторных передач в клетке вызывают:
1) вирус группа;
2) холерный токсин;
3) токсин кишечной палочки;
4) экзотоксин коклюша.
14. Назовите клеточного адаптационные механизмы метаболическо-функционального характера:
1) снижение функциональной активации клеток;
2) активация синтеза белков теплового шока;
3) регенерация;
4) гиперплазия.
15. Основными механизмами компенсации расстройств регуляции внутриклеточных процессов являются:
1) активация антиоксидантной системы;
2) угнетение гликолиза;
3) изменение чувствительности мембранных рецепторов к сигнальным молекулам;
4) изменение активности аденилатциклазы.
16. Укажите уровни реализации межклеточных механизмов адаптации:
1) клеточный;
2) органно-тканевой;
3) интегративный;
4) внутрисистемный.
17. Различают следующие стадии апоптоза:
1) стадия инициации;
2) стадия трансмембранного переноса сигнала апоптоза;
3) стадия репрессии генов, контролирующих синтез белков – ингибиторов апоптоза;
4) стадия реализации программы апоптоза.
18. К заболевания, связанных с усилением апоптоза относят:
1) лимфома Беркита;
2) токсические гепатиты;
3) болезнь Альцгеймера;
4) системная красная волчанка.
19. Задачами патогенетической терапии клетки являются:
1) устранение (уменьшение) нарушений энергообеспечения клетки;
2) предупреждение (уменьшение) прогрессирования повреждения мембраны клетки;
3) коррекция водно-солевого гомеостаза клетки;
4) устранение недостатка (избытка) инградиентов пищи, способствующих развитию патологии клетки.
СИТУАЦИОННЫЕ ЗАДАЧИ
Ситуационная задача № 1
В клинику поступил больной К., 45 лет с жалобами на головные боли, мышечную слабость, судороги. АД – 170/110 мм рт. ст.
При обследовании обнаружено: полиурия (повышенное суточное выделение мочи), гипостенурия (выделение мочи постоянно низкого удельного веса), снижение концентрации калия в крови (гипокалиемия), увеличение количества натрия в крови (гипернатриемия), повышение содержания 17-оксикортикоидов в моче.
Вопросы:
1. Назовите и обоснуйте предполагаемую причину повышения уровня артериального давления у пациента.
2. Какие дополнительные исследования еще необходимо провести для окончательного установления причины развития артериальной гипертензии?
3. Объясните механизм развития артериальной гипертензии и клинических проявлений у данного больного.
4. Нарушением какой составляющей гомеостаза клетки вызвано заболевание у пациента?
Ситуационная задача № 2
60 лет поступил в клинику с жалобами на интенсивные боли за грудиной, сохраняющиеся в течение 2,5 часов. Со слов больного перед этим, в течение недели находился в эмоциональном напряжении, много курил.
Объективно: кожные покровы бледные, акроционоз. Частота дыхания – 26 в мин, хрипов нет. тоны сердца приглушены, аритмичны, АД – 95/75 мм рт. ст. Данные ЭКГ: периодическая мерцательная аритмия предсердий с частотой 350 импульсов в минуту, блокада проведения импульсов в правой ножке Гисса, подъем сегмента ST в I, AVL, V1 – V4 отведениях. Был выставлен диагноз: острый инфаркт миокарда, сердечная аритмия, сердечная недостаточность.
Вопросы:
1. Назовите и обоснуйте возможную причину развития инфаркта (некроза) миокарда у данного больного.
2. Охарактеризуйте звенья патогенеза структурно-функциональных нарушений кардиомиоцитов и укажите их связь с клиническими проявлениями у данного больного.
3. Нарушением какой составляющей гомеостаза клетки (кардиомиоцита) вызвано данная патология?
11. ЭТАЛОНЫ ОТВЕТОВ
Эталоны ответов на тестовое задание: 1. 2); 2. 3); 3. 2),4),5); 4. 1),2),4); 5. 1),2); 6. 1),3); 7. 1),4); 8. 3),4); 9.1),3); 10. 1),4); 11. 1),4); 12. 1),2); 13. 2),4); 14. 1),2); 15. 3),4); 16. 2),4); 17. 1),4); 18. 2),3); 19. 1),3).
Эталоны ответов к задаче №1:
1. У данного больного вероятной причиной артериальной гипертензии является нарушение водно-электролитного обмена (гипокалиемия, гипернатремия). В пользу этого свидетельствует и увеличение в моче концентрации 17-оксикортикостероидов – продуктов метаболизма кортикоидов – гормонов коры надпочечников (они, в частности альдостерон, активно регулируют содержание Nа+ и К+ в организме).
2. Для установления точной причины необходимо провести рентгенографию (или УЗИ) надпочечников и определить концентрацию альдостерона в крови. УЗИ (ультразвуковое исследование) – выявило опухолевидное образование (0,5х0,5 см) в коре правого надпочечника. В крови – повышенное содержание альдостерона. Заключение – гормонпродуцирующая опухоль коры надпочечников – альдостерома.
3. Избыток альостерона усиливает реабсорбцию Nа+ и стимулирует экскрецию К+ эпителием почечных канальцев. Натрий (осмотически активное вещество) задерживает воду в организме, увеличивается объем циркулирующей крови, причем она гиперосмолярна (гиперосмия). Последнее активирует осморецепторы, что стимулирует секрецию антидиуретического гормона (АДГ, продукт гипоталамуса). АДГ повышает реабсорбцию Н2О в дистальных канальцах и собирательных трубочках нефрона – ОЦК еще большей степени возрастает. Увеличение объема циркулирующей крови – это не единственный механизм повышения уровня АД. Другой – обеспечивает гиперосмия: увеличение концентрации Nа+ повышает чувствительность рецепторов мышечных элементов артериол к вазопрессорным агентам (например, катехоламинам).
Дисбаланс в концентрациях Nа+ и К+ приводит к нарушению электрогенза в мышечных клетках и расстройству нервно-мышечного возбуждения. Об этом сидетельствует наличие у данного больного мышечной слабости и судорог. Гипостенурия является следствием низкого содержания Nа+ в моче.
4. У данного пациента причиной развития патологии послужило нарушение информационной составляющей гомеостаза клеток. Избыток альдостерона (сигнальная молекула) «заставил» клетки (почек) длительное время работать, эксплуатировать программы, не отвечающие потребностям организма. Возникшая вследствие этого сложная цепь причинно-следственных отношений и проявилась в виде клинических симптомов.
Эталоны ответов к задаче №2:
1. Вероятной причиной развития инфаркта (некроза) миокарда у данного больного послужила гипоксия, возникшая вследствие нарушения коронарного кровотока. В пользу этого свидетельствует предшествующее длительное эмоциональное возбуждение. Оно характеризуется активацией симпато-адреналовой системы – спазм коронарных сосудов, который может усиливаться на фоне присутствия фактора риска (табакокурение). Нельзя исключить и поражение коронарных сосудов атеросклеротическим процессом (больному 60 лет). При этой патологии создаются благоприятные условия для тромбообразования с последующим закрытием тромбом просвета сосудов миокарда.
2. Существенное значение в патогенезе повреждения гипоксией кардиомиоцитов (вплоть до их некроза) является энергодефицит. Это можно представить в виде развития следующих, причинно-следственных связей: дефицит АТФ – затруднение работы К+/Nа+ ионного насоса – выход К+ из клетки, накопление Nа+ в клетке – сглаживание калий-натриевого градиента клетки – изменение электровозбудимости кардиомиоцита, нарушение проведение импульсов возбуждения, формирование патологических очагов возбуждения. О наличии данных нарушений свидетельствуют результаты анализа ЭКГ – мерцательная аритмия, блокада правой ножки Гисса и другие отклонения электрофизиологических параметров миокарда.
Эти метаболические изменения, а так же ряд других характерных нарушений свойственных гипоксии (внутриклеточный ацидоз, активация перекисного окисления жиров и др.) значительно угнетают функцию сердечной мышцы – формируют сердечную недостаточность. О ее наличии косвенно свидетельствуют: бледность кожных покровов, акроцианоз, снижение уровня артериального давления и болевой синдром. Появление последнего по всей вероятности связано с накопление в очаге поражения продуктов измененных метаболических процессов (например, ионов Н+ из-за развития ацидоза).
3. В основе развития инфаркта миокарда у данного больного дежат изменения метаболической составляющей гомеостаза кардиомиоцита. Недостаток кислорода вызвал нарушение работы митохондрий мышечных клеток сердца, что способствовало развитию энергодефицита, ионному дисбалансу, формированию внутриклеточного ацидоза и др.
12. ЛИТЕРАТУРА
1. Введение в молекулярную медицину / Под редакцией . – М.: «Медицина», 2004. – 496 с.
2. , , и др. Роль лизосомальных ферментов в генезе ведущих клинико-патофизиологических синдромов: факты и гипотезы // Пат. физиол. и экспер. терапия. – 2007. – №1. – С. 18-21
3. , Чурилов общей патологии. Часть I. Основы общей патофизиологии (Учебное пособие для мед. вузов). – СПб, ЭЛБИ, 1999. – 624 с.
4. Литвицкий : Учебник: 4-е изд. перераб. и доп. – М.: ГЭОТАР-Медиа, – 2007. – 496 с.
5. , Кузнецов биология (Учебное пособие для мед. вузов). – М.: информационное агентство», 2003. – 544 с.
6. , , и др. Модуляция апоптоза мононуклеаров в условиях окислительного стресса // Бюлл. экспер. биол. и мед. – 2008. – Т.145, №3. – С. 251-254.
7. Общая патология: Учебное пособие для студ. мед. вузов / Под редакцией . – М.: Издательский центр «Академия», 2006. – 336 с.
8. , Архипенко баланса прооксидантов и антиоксидантов – равнозначных участников метаболизма // Пат. физиол. и экспер. терапия. – 2007. – №3. – С. 2-18
9. , Молекулярная биология клетки. Руководство для врачей. Пер. с англ. – М.: БИНОМ-Пресс, 2004. – 272 с.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
