

Цитоскелет, помимо опорной и локомоторной функции, осуществляет и внутриклеточное перемещение органоидов, включений, секреторных гранул. Обеспечивает прикрепление клеток друг к другу (с помощью десмосом) и межклеточному веществу, участвует в передаче сигнала от мембранных рецепторов внутрь клетки.
Нарушение функции цитоскелета может быть следствием:
· энергодефицита, так как он совершает свою механическую работу за счет расщепления АТФ и ГТФ. Наблюдается угнетение актинмиозиновой (в микрофиламентах) или тубулин-динеиновой (в микротрубочках) скользящих систем. Например, при сахарном диабете развивается синдром «ленивых фагоцитов», характеризующийся замедлением хемотаксиса и снижением фагоцитарной активности данных клеток. И происходит это, как раз из-за нарушения энергообразования (уменьшается поступление глюкозы в клетки). В результате – течение сахарного диабета осложняется иммунодефицитом.
Значительные нарушения цитосклелета наблюдаются при выраженной гипоксии, отмечающееся при этом набухание клеток, сопровождается отсоединением плазматической мембраны от элементов цитоскелета. Например, острая ишемия миокарда характеризуются отсоединением сарколеммы кардиомиоцитов от промежуточных филаментов. В результате снижается механическая плотность клеток;
· нарушения полимеризации и деполимеризации компонентов цитоскелета. Они могут быть наследственными, как например, при синдроме Чедиака-Хигаши. Он характеризуется нарушением полимеризации микротрубочек цитоскелета, отсюда, замедление слияния фагосом с лизосомами в фагоцитах и угнетение киллерного эффекта НК-лимфоцитов (натуральных киллеров). Клинически синдром проявляется частыми и длительными инфекционными заболеваниями, наиболее часто гноеродной природы; нарушением хемотаксиса лейкоцитов и их выхода из костного мозга. Неврологическая симптоматика (нистагм, умственная отсталость, периферическая нейропатия) сопровождающая развитие синдрома, может быть объяснена так же дефектами цитоскелета нейронов.
Приобретенные нарушения полимеризации и деполиризации цитоскелета встречаются чаще. Есть ряд токсинов, избирательно повреждающих цитоскелет. Цитохалазины вызывают деполимеризацию, а фаллодин (токсин бледной поганки) – полимеризацию актина. Колхицин угнетает полимеризацию, а таксол – деполимеризацию микротрубочек. При злокачественной трансформации клетки, один из онкобелков вызывает необратимое фосфорилирование цитоскелетного белка винкулина (он в норме принимает участие в прикреплении клетки к межклеточному веществу). Поэтому злокачественные клетки свободно отсоединяются от межклеточного вещества и мигрируют в другие органы и ткани. Это считается одним из важных механизмов способности опухолевых клеток к метастазированию;
· структурных нарушений, что характерно при поражении клеток рядом вирусов. Например, реовирусы (оспенный вирус и др.) взаимодействуют непосредственно со структурами цитоскелета. Они способны вызывать разрыв винтиновых промежуточных филаментов, изменения тубулина микротрубочек и слияние клеток. В результате действия данных вирусов может отмечаться угнетение функции ресничек дыхательного эпителия (нарушается работа мукоцилиарного клиренса), активности фагоцитов и образование многоядерных гигантских клеток;
· формирования иммунопатологических механизмов. При этом виде повреждения цитоскелета большое значение имеют выше указанные вирусы. Они содержат специфические рецепторы к белкам цитоскелета. Формирующийся организмом иммунный ответ против вырусных антигенов может сопровождаться появлением аутоантител, копирующих способность вируса связываться (реагировать) с элементами цитоскелета. В связи с этим, многие вирус-индуцированные заболевания продолжаются как аутоиммунные, т. е. сопровождаются появлением аутоантител к фрагментам цитоскелета. Например, вирусный гепатит С. Он инициируется данным вирусом, но его дальнейшее, волнообразное течение поддерживается синтезом аутоантител к белкам цитоскелета – кератину и актину.
Одной из причин бесплодия может быть наличие аутоантител (особенно их высокие концентрации) к элементам цитоскелета сперматозоидов (резко снижается их подвижность). Установлено, что титр антиспермальных антител у женщин повышается при беспорядочных половых связях с различными партнерами (в норме титр незначителенчто титр антиспермальных антител у женщин повышается при беспорядочных половых связях с различными партнерами ()м ирующих спосо). По всей видимости такой стиль половой жизни, из-за иммунизации более широким кругом различных спермальных антигенов, и приводит к активной выработки женским организмом антител к сперматозоидам.
К типовым клеточным изменениям относят отложение гиалина (гиалиновая диспротеинемия) – гомогенной прозрачной белковой массы. В качестве источника внутриклеточного гиалина может быть агрегация промежуточных филаментов (белок прекератин). Они, кстати, являются наиболее ранимыми элементами цитоскелета.
Подобные агрегации нередко являются следствием описанных выше нарушений и возникают под влиянием разнообразных причин. Наблюдаются при алкогольном поражении печени, болезни Коновалова-Вильсона (гепатоциты повреждаются избытком меди), тяжелом ожирении, сахарном диабете и др.
Таким образом, наиболее яркими, имеющих большое патогенетическое значение, проявлениями нарушения функции цитоскелета является угнетение фагоцитоза, пиноцитоза и хемотаксиса. порме от незначителен
5. ХАРАКТЕРНЫЕ ИЗМЕНЕНИЯ В ЦИТОЗОЛИ.
Цитозоль (гиалоплазма, цитоплазма) – выполняет роль депо, где осуществляется накопление гликогена, липидов (триглицириды и эфиры холестерина). Последние используются как источник энергии и для синтеза стероидных гормонов, гликоген – запас (источник) глюкозы. В гиалоплазме находится до 40% белков клетки, основное количество внутриклеточной воды (свободная, мобильная) тысячи ферментов, осуществляющих синтез и распад белков, жиров и углеводов. Многообразие функций клетки обеспечивается пространственной и временной регуляцией определенных метаболических путей. Пространственная регуляция связана со строгой локализацией ферментов в различных органеллах (компартментализация). Наличие определенного количества осмотически активных веществ (в виде анионов и катионов) и ионов водорода играют важную роль в функционировании клетки, и в первую очередь, за счет поддержания в ней оптимальных значений рН, осмотического давления.
Как мы уже отмечали (см. раздел механоосмотическое растяжение и разрыв мембраны) накопление Nа+ и Са++ в клетке способствует увеличению ее осмотического давления и развитию внутриклеточной гипергидратации (по закону осмоса Н2О перемещается в зону повышенного осмотического давления).
Внутриклеточная гипогидратация (обезвоживание) чаще всего формируется при увеличении осмолярности внеклеточной жидкости, т. е. без структурных нарушений клетки (работает все тот же закон осмоса). Кстати, и гипергидратация клетки может наблюдаться тоже без нарушения проницаемости мембраны и работы ионных насосов – при уменьшении осмолярности внутриклеточной жидкости. Более подробно см. в разделе «Патофизиология водно-минерального обмена».
Патогенетической основой клинических симптомов гипергидратации клеток является набухание и разрыв мембран (осмотический гемолиз эритроцитов) и различные функциональные нарушения. Например, «набухание» нейронов ЦНС – головная боль, апатия, нарушение сознания и др. Гипогидратация клеток сопровождается сморщиванием ядра, уменьшением объема органелл, вплоть до их распада. Наиболее ярко это проявляется в нейронах и сопровождается следующими клиническими симптомами - возбуждением, беспокойством, комой и др.
Значения рН гиалоплазмы зависят от процессов анаэробного гликолиза, пентозного цикла и микросомального окисления. Анаэробный гликолиз активируется при всех видах гипоксий и ослаблении функции митохондрий, т. е. при угнетении основного пути (аэробного) получения АТФ. Возрастание роли анаэробного гликолиза в синтезе энергетических веществ сопровождается накоплением недоокисленных продуктов белка – молочной, пировиноградной кислоты и др., а это способствует возникновению внутриклеточного ацидоза.
Избыток Н+ (низкое значение рН цитоплазмы) угнетает активность внутриклеточных ферментов, цитоскелета, ряда органелл. Это существенно отражается на различных функциональных возможностях клеток – их сокращении, секреции, эндоцитоза и др.
Внутриклеточный ацидоз, гипо - и гипергидратация клетки, гипоксия и др. могут приводить к нарушению упорядочного характера циркуляции цитоплазматической жидкости (т. е. изменять ее вязкость) и влиять на компартментализацию ферментов. Например, при внутриклеточной гипоксии замедляется движение нейтрофилов и одним из механизмов этого – медленное перемещение гиалоплазмы (из-за повышение ее вязкости) в псевдоподии данных клеток. Примером нарушения внутриклеточной локализации ферментов может служить появление в цитоплазме гидролитических энзимов лизосом, цикла Кребса (в норме они находятся в лизосомах и матриксе митохондрий) и др.
В нормальных клетках, ее цитозоле, встречаются различные включения (гликоген, липиды и др.). При патологии данные образования могут претерпевать качественные и количественные изменения – их рассматривают как одну из типовых форм повреждения клетки – дистрофия. Они подробно изучаются на курсе патологической анатомии.
Мы закончили рассмотрение механизмов нарушений метаболической составляющей гомеостаза клетки и приступаем к характеристики патологии информационных процессов (информационной составляющей гомеостаза клетки).
6. НАРУШЕНИЕ ИНФОРМАЦИОННЫХ ПРОЦЕССОВ В КЛЕТКЕ (нарушения информационной составляющей гомеостаза клетки).
На рис. 11 приведена общая схема информационных нарушений, которые могут обуславливать развитие болезней. Информация клеткам представляется в виде различных химических регуляторных сигналов (управляющие агенты, сигнальные молекулы, первичные мессенджеры). Они получили название «лигандов» (от лат. ligare – связывать), для них, на поверхности клеток существуют специфические рецепторы (см. выше). После взаимодействия лиганда с соответствующим рецептором, информация, с помощью пострецепторных посредниковых механизмов (пострецепторный передатчик), доставляется в генетический аппарат или ключевым ферментам и структурным белкам цитоплазмы. В результате этого и вызывается (возникает) тот конечный эффект (ответ), который «требовал» от клетки лиганд:
· изменяется активность соответствующих ферментов или структурных белков цитоплазмы;
· изменяется активность соответствующих генов и скорость синтеза ферментов или структурных белков.
То есть, соответствующая адаптационная программа на действие сигнальной молекулы включается, и клетка может нормально функционировать в изменившихся условиях.
Возникновение заболеваний может наступить в результате сбоев в работе на каждом из этих этапов информационных взаимоотношений (рис. 11):
· качественные и количественные нарушения управляющих агентов (патология сигнализации);
· нарушения рецепции сигналов;
· нарушения функционирования пострецепторных посредниковых механизмов (пострецепторного передатчика);
· дефекты клеточных адаптационных программ.
 |
 |
 |

 |
 |
Рис.11. Типы информационных нарушений, лежащих в основе болезней. Клетки - программные системы, дающие адаптивные ответы в рамках генетических стереотипов. Болезнь наступает из-за нарушения сигнализации, рецепции, пострецепторного сопряжения, работы исполнительного аппарата и дефектов программы. Ошибки программы – технические дефекты, несоответствие программы ситуации – технологические дефекты (по , 1999г.).
Патология сигнализации. Все химические регуляторные вещества (сигналы) распределены на следующие группы: гормоны, медиаторы, антитела, субстраты и ионы. Причиной заболевания может быть избыток, недостаток и мимикрия (от англ. mimicry – подражание, маскировка) сигнала (ошибочное восприятие одного сигнала вместо другого).
Избыток управляющего сигнала. Он заставляет клетку излишне интенсивно или длительно функционировать. Например, повышенное содержание в крови глюкокортикоидов (синдром Иценко-Кушинга) вынуждает клетки усиленно эксплуатировать программы метаболической регуляции. Вследствие этого усиливается липогенез и глюконеогенез, развивается отрицательный азотистый баланс, метаболический алкалоз. Могут даже стимулироваться механизмы клеточной гибели (апоптоз), что приведет, например, к иммунодефициту (гибель лимфоидных клеток). Повышение титра аутоантител инициирует развитие аутоиммунных заболеваний, хотя их низкие концентрации отмечаются у совершенно здоровых людей, в норме они участвуют в регуляции роста и функций клеток.
Недостаток управляющего сигнала. Отсутствие или нехватка сигнальных молекул характеризуется тем, что клетка не может активировать ту или иную программу ответа, необходимую для ее нормальной жизнедеятельности в конкретной ситуации. Например, при снижении синтеза инсулина поджелудочной железой уменьшается поступление глюкозы в инсулинзависимые органы (инсулинзависимый сахарный диабет). Недостаток белка (управляющий агент – субстрат) способствует развитию «квашиор» – заболеванию, вызванному дефицитом экзогенного белка и проявляющегося задержкой роста, гипопротеинемией, жировым перерождения печени и др.
Мимикрия управляющего сигнала. Возникает при ситуациях, когда клеточный рецептор, отвечающий за активацию той или иной программы «ошибочно» реагирует не со «своей» сигнальной молекулой. Наиболее часто мимикрия связана с выработкой аутоантител, иммунологически копирующих ряд гормонов или медиаторов и способных реагировать с их рецепторами («иммунологический имидж» сигнала). Например, Базедова болезнь (диффузный токсический зоб) характеризуется усиленным синтезом гормонов щитовидной железы. Нередко гиперфункция железы объясняется не активирующим влиянием на нее физиологического стимулятора – тиреотропного гормона (сигнальная молекула – ТТГ), а его иммунологической копии – LАТS (длительно действующий стимулятор щитовидной железы). LАТS – аутоантитело (IgG) к рецепторам для ТТГ, при взаимодействии с которыми, тиреоциты повышают свою активность. Происходит это на фоне нормальной или даже сниженной концентрации тиреотропного гормона гипофиза в крови у данных больных. Аминокислотный дисбаланс (при печеночной недостаточности) приводит к синтезу ложных нейротронсмиттеров (сигнальные молекулы в ЦНС) – β-фенилэтиламина и октопамина. По структуре они сходны с допамином и норадреналином (истинные нейротрансмиттеры), но значительно превосходят их в активности. Отсюда, вытесняя истинные лиганды с их рецепторов, ложные сигнальные молекулы блокируют постсинаптическую передачу, что ведет к развитию патологии (извращения сна и бодрствования, хлопающему тремору и др.).
Отсутствие патологии сигнализации не всегда гарантирует ответ клетки должным образом, и, одной из причин этого, может служить нарушение восприятия рецепторами клетки своих управляющих агентов.
Патология рецепции сигналов. Нарушения данного звена передачи информации объясняется:
· увеличением или уменьшением количества рецепторов;
· изменением чувствительности рецепторов;
· нарушениями конформации рецепторных макромолекул.
Они могут быть наследственными и приобретенными. В качестве примера наследственной недостаточности рецепторов можно привести семейную наследственную гиперхолестеринемию. Ее патогенез связан с дефектом рецептора, отвечающего за распознавание клетками эндотелия сосудов белкового компонента липопротеидов низкой (ЛПНП) и очень низкой плотности (ЛПОНП). В норме, с помощью данного рецептора (апопротеина В):
· регулируется поступление ЛПНП и ЛПОНП в клетки кровеносных сосудов;
· предотвращается их перегрузка холестерином, снижается синтез собственного холестерина, активируется его этерификация и увеличивается выведение холестерина из клетки.
При дефекте гена, контролирующего синтез апопротеина В, холестеринсодержащие вещества все равно поступают в клетку. Однако, выше описанная защитная метаболическая программа практически не работает; в клетке накапливается холестерин, и, в конечном итоге формируется картина атеросклеротического поражения кровеносных сосудов.
Приобретенная патология клеточных рецепторов наблюдается значительно чаще наследственной. Известны различные химические соединения (лиганды-антогонисты) препятствующие взаимодействию с рецепторами «своих» управляющих агентов. Например, у некоторых больных с гипо - и апластическими анемиями выявляются антитела к рецепторам стволовых клеток. Значительно изменяется характеристика клеточных рецепторов при нарушении структуры липидного слоя мембраны клеток (см. выше).
Патология пострецепторных передаточных механизмов. Нормальное функционирование двух первых этапов доставки информации еще не дают возможности клетки включать ту или иную адаптационную программу. Местом их инициации является ядро или цитоплазма, куда и доставляется управляющий сигнал с помощью каскадного механизма ферментативных реакций.
В зависимости от полярных свойств управляющих агентов, они подразделяются на две группы:
· полярные или гидрофильные сигнальные молекулы – белки, пептиды, производные аминокислот (кроме тиреоидных гормонов). Они не растворяются в жирах.
· неполярные или гидрофобные сигнальные молекулы – стероиды, производные жирных кислот, тиреоидные гормоны. Жирорастворимы.
Данное разделение первичных мессенджеров имеет принципиальное значение и связано в первую очередь с механизмами их действия на клетку-мишень.
Для каждой сигнальной молекулы, не растворяющейся в жирах, имеется свой мембранный рецептор (R, рис. 12). Возбуждение рецептора соответствующим лигандом ведет к изменению концентрации в клетке определенного внутриклеточного посредника (вторичного месенджера, Х, рис. 12).
Гормон
 |
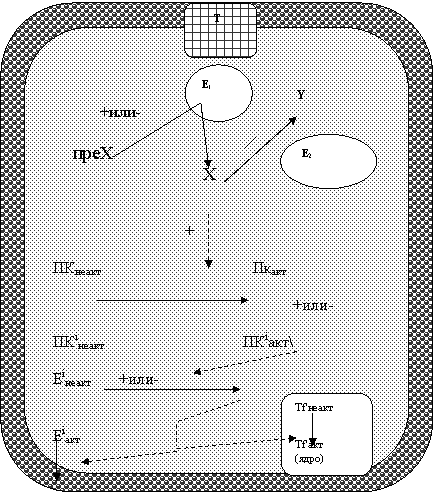 |
Рис. 12. Общая схема действия полярных (гидрофильных) гормонов
В настоящее время наиболее изученными из них являются: циклический аденозинмонофосфат (ц. АМФ), циклический гуанозинмонофосфат (ц. ГМФ), диацилглицерол (ДАГ), инозитолтрифосфат (ИТФ), Са2+, Rаs-белок и др. Концентрация вторичных мессенджеров определяется активностью ключевых ферментов их образования (Е1) или инактивации (Е2) (рис. 12). Е1 и Е2 находятся под мембраной (мембраносвязанные белки, периферические белки). Поэтому, возбуждение рецепторов должно сказываться на активности одного из них, что часто (но не всегда) осуществляется с помощью трансмембранного белка-трансмиттера (Т, рис. 12), передающего сигнал от рецептора на фермент Е1 или Е2.
Дальнейший ход событий рассмотрим на примере образования возбуждающего фермента (Е1). В зависимости от специфики сигнальной молекулы активируются различные Е1. Например, для увеличения ц. АМФ необходима активация аденилатциклазы (АЦ). Гуанилатциклаза повышает активность ц. ГМФ.
В роли белков-трансмиттеров выступают различные соединения, к наиболее из них известных относятся белки класса-G.
Вторичный посредник (Х) в свою очередь повышает активность той или иной протеинкиназы (ПК). Например, ц. АМФ активирует Пк типа А, ц. ГМФ – Пк типаG. Протеинкиназы – это специальные регуляторные ферменты, которые за счет фосфорилирования строго определенных белков, в конечном счете и определяют ответ клетки (включение той или иной адаптационной программы). Они изменяют:
· активность соответствующих ферментов или структурных белков (Еi);
· активность соответствующих генов и скорость синтеза ферментов или структурных белков (Тfi).
В регуляторной цепочке нередко имеется не одна ПК, а каскад из двух (ПК→ПКi) и более протеинкиназ. Активированные белки (путем фосфорилирования) по мере необходимости инактивируются дефосфолированием (протеинфосфотазами). Т. е. фосфорилирование и дефосфорилирование – один из наиболее универсальных способов регуляции активности белков – как структурных, так и ферментов.
Для гидрофобных (липофильных) сигнальных молекул мембранные рецепторы не требуются – управляющие агенты легко диффундируют через мембрану клетки-мишени. В цитоплазме (или ядре) для них содержатся специфические рецепторные белки. Комплекс рецепторов – сигнальная молекула влияет на активность тех или иных генов, тем самым повышая синтез определенных белков.
Мы рассмотрели общую схему механизмов постецепторной передачи и информации клетке в норме. На каждом из этих этапов могут возникать нарушения, они и будут предметом дальнейшего изложения материала.
Клинико-патофизиологическая характеристика нарушений пострецепторных передач:
· повреждение трансмембранного белка-трансмиттера (Т, рис. 12). Из этого класса белков наиболее известна патология так называемых G-белков, состоящих из трех основных субъединиц. При наследственной остеодистрифии Олбрайта в результате мутации одного из белков G (GаS) прерывается передача сигнала от Т к Е1 (Е1-аденилатциклаза). Типичными проявлениями данного состояния служат рассеянные очаги разряжения костей скелета, гипоплазия зубной эмали и др. Нередко нарушения на этом этапе следования сигнала отмечаются при инфекционной патологии. Так, холерный токсин способствует длительному активному состоянию Gs, что приводит к продолжительной экскреции воды и электролитов клетками кишечного эпителия. Отсюда – диарея (понос) и водно-электролитные нарушения. Экзотоксины бордетеллы (коклюша) действуя аналогичным образом в клетках эпителия бронхов, вызывают кашель, снижают бактерицидную активность лейкоцитов. Повышенная активность G-белков, например в клетках эндокринной системы, может служить митогенным стимулом (через активацию ц. АМФ), что повышает риск злокачественных новообразований;
· изменение активности ферментов образования и инактивации вторичных мессенджеров (Е1, Е2, рис. 12) . На этом этапе пострецепторных механизмов информация может изменяться под влиянием различных химических соединений. К примеру, токсин сибирской язвы, обладая аденилатциклазной активностью, вызывает отек кожи (при кожном пути заражения) или понос (при кишечном пути заражения). Аналогичный аденилатциклазный механизм свойственен и эндотоксину коклюша (помимо выше указанного его влияния на G-белки);
· изменения активности вторичных посредников (Х) и протеинкиназ (ПК). Концентрация вторичных мессенджеров (а следовательно и их активность), как правило находится в прямой зависимости от наличия ферментов Е1 или Е2. В качестве примера можно привести эффект действия кодеина. Помимо прочих механизмов, кодеин ингибирует фосфодиэстэразу, которая снижает концентрацию ц. АМФ в клетке. Следствием угнетения активности фосфодиэстеразы будет повышение концентрации ц. АМФ, результат – повышения активности клеток. Это наглядно проявляется в работе нейронов коры головного мозга – увеличивается память, скорость ориентировочных реакций и др. Однако, длительная стимуляция данным препаратом, острое отравление приводит к многочисленным нарушениям высшей нервной деятельности и других органов и систем. Так, появляется немотивированное беспокойство, тремор, нарушения нормального цикла сна и др.
Первичные изменения протеинкиназ (без нарушений предшествующих путей передачи сигнала) можно продемонстрировать на примере бластной трансформации клетки. В норме, один из путей передачи сигнала к митозу клетки опосредуется Rаs-белком (вторичный мессенджер). Он, в активном состоянии, запускает целый каскад митогенактивирующих протеинкиназ (МАПК). МАПК, модифицируя соответствующие транскрипционные факторы (Тf', рис.12), способствует активации генов митоза и пролиферации клеток. Здоровые клетки без специфического лиганда (обычно это ростковые факторы) не размножаются. При мутации гена, отвечающего за синтез того или иного белка-фермента в системе МАПК, например Rаf-протеинкиназы, управляющего сигнала уже не нужно. Дело в том, что мутация может вызвать длительную гиперэкспрессию данного гена, позволяющую Rаf-протеинкиназе длительно, и не зависимо от «указаний свыше» сохранять повышенную активность. Клетки вовлекаются в бесконечную, неконтролируемую организмом серию делений, что рассматривается в настоящее время как один из этапов их озлакачествления.
На этом мы закончим рассмотрение нарушений посрецепторных информационных механизмов в клетке. Мы не касались еще очень многих других путей передачи информации, например, таких вторичных мессенджеров как инозиттрифосфат (ИТФ) и диацилглицерин (ДАГ), конечный эффект которых складывается из эффектов действия протеинкиназы С и ионов Са++. Но даже приведенные выше примеры свидетельствуют о большом значении неадекватного ответа клетки в развитии болезней при «сбоях» в постерцепторных механизмах.
Программа, не соответствующая ситуации (технологический дефект). Многие адаптационные программы при различных патологических процессах адекватно реагируют на управляющие агенты. Но и здесь есть проблемы. К сожалению не всегда, казалось бы соответствующая защитная реакция организма на воздействие патогенного агента, обладает абсолютной «полезностью». Речь идет об их относительной целесообразности и потенциальной патогенности, о своеобразном технологическом дефекте адаптационных программ (технологическом несовершенстве). Например, совершенно очевидно положительное значение формирование отека в очаге воспаления (разведение токсических продуктов, задержка их в месте образования и др.). В тоже время просматривается и его негативные стороны – сдавление экссудатом кровеносных сосудов, развитие гипоксии, и при определенных условиях, это может послужить утяжелению патологического процесса (эндогенезации). Данный вопрос мы подробно рассматривали, и что бы не повторяться, рекомендуем обращаться к учебному пособию «Патофизиология: вопросы общей нозологии» (, 2004).
Технические дефекты адаптационных программ. В данном случае мы говорим о дефектах информации, содержащейся в ДНК (технические ошибки в записи клеточных адаптационных программ). В основе этих нарушений лежат половые мутации (см. выше).
Клинико-патофизиологическая характеристика. Половые мутации определяют развитие наследственных заболеваний, то есть главным звеном патогенеза которых служит первичный технический дефект в программном аппарате клетки. Например, возникновение фенилкетонурии объясняется дефектом ответа клеточной программы гепатоцита на фенилаланин (дефект гена отвечающего за синтез фермента фенилаланин-4-гидроксилазы). Недостаток данного фермента замедляет скорость превращения фенилаланина в тирозин и приводит к резкому увеличению его концентрации в крови больного. Нарушение обмена фенилаланина провоцирует еще ряд метаболических изменений, что в итоге и определяет становление и симптоматику фенилкетонурии – «осветление» кожи, глаз и волос (дефицит меланина), снижение уровня артериального давления (нарушение обмена катехоламинов), снижение интеллекта (токсическое действие на мозг метаболитов фенилалнина, например, фенилэтиламина и др.).
Мы завершили изучение различных нарушений клетки, возникающих при ее взаимодействии с патогенным агентом или являющихся следствием нарушений информационных процессов. Степень их выраженности, вероятность развития необратимых последствий (рис. 1, т. необратимости) с последующим развитием некроза, во многом определяется состоянием защитно-приспособительных механизмов клетки. Следовательно, мы переходим к изучению второй составляющей паранекроза клетки – адаптации клетки к повреждению.
7. МЕХАНИЗМЫ АДАПТАЦИИ КЛЕТКИ
Выше было отмечено значение защитно-приспособительных механизмов как в норме, так и при патологии. Ответ клетки на воздействие этиологического фактора в виде паранекроза становиться возможным при их недостаточности, но и здесь роль данных механизмов велика. Они уменьшают степень повреждения клетки и их последствий, при определенных обстоятельствах (например, ликвидация патогенного агента) способствуют возвращению ее к исходному состоянию. Однако необходимо помнить, что механизмы адаптации, в силу своей относительной патогенности, могут вызывать вторичные повреждения (эндогенезация патологического процесса).
Большое многообразие механизмов адаптации клеток к повреждению можно систематизировать следующим образом:
I. Внутриклеточные механизмы адаптации
1.Защитно-приспособительные механизмы метаболическо-функциональ-ного характера. Они направлены на:
· компенсацию нарушений энергообмена клеток;
· защиту клеточных мембран и ферментов;
· устранение или уменьшение нарушений обмена воды и электролитов клетки;
· компенсацию расстройств механизмов регуляции внутриклеточных процессов, в том числе и их первичных нарушений (информационной составляющей гемостаза);
· ликвидацию дефектов генетического аппарата (сохранение генетических программ) клетки;
· активацию синтеза белков теплового шока (БТШ, HSP);
· снижение функциональной активности клеток.
Данные механизмы можно отнести к механизмам срочной компенсации, эффект большинства из них проявляется сравнительно быстро, они являются своеобразной «первой линией защиты».
2. Защитно-приспособительные механизмы морфологического характера. К ним относят – регенерации, гипертрофии и гиперплазии. Они формируются при длительном или периодическом воздействии патогенного фактора и обеспечивают долговременную адаптацию клеток за счет регенерации, гипертрофии и гиперплазии.
II. Межклеточные (системные) механизмы адаптации.
По уровню их реализации выделяют:
· органно-тканевые;
· внутрисистемные;
· межсистемные.
Внутриклеточные механизмы адаптации
1. Защитно-приспособительные механизмы функционально-метаболи-ческого плана.
Компенсация нарушений энергообмена клеток. Обязательным условием успешной работы практически всех механизмов клеточной адаптации является их достаточное энергетическое обеспечение. Поэтому восстановление энергетического баланса клеток, повышение его ресурсов имеет первостепенное значение и это достигается следующим образом:
· активируется ресинтез АТФ в сохранившихся митохондриях, а так же и за счет активации гликолиза. Интенсивность анаэробного гликолиза может возрастать до 15-20 раз (в сравнении с нормой). При слабых и умеренных повреждениях повышается активность ферментов окислительного фосфорилирования, увеличивается сродство к кислороду;
· активируются механизмы транспорта энергии. Например, возрастает активность креатинфосфокиназы, адениннуклеотидтрансферазы;
· усиливается эффективность ферментов утилизации энергии, в частности, аденозинтрифосфотазы.
Защита клеточных мембран и ферментов. Она осуществляется за счет:
· активации антиоксидантной системы (см. выше);
· активации синтеза, упаковки и доставки компонентов плазмолеммы вместо (взамен) ее поврежденных участков (эндоплазматический ретикулум, аппарат Гольджи);
· активации процессов внутриклеточной детоксикации. Центральным местом в клетке, где происходит обезвреживание различных токсических веществ является гладкий эндоплазматический ретикулум. В его мембранах локализованы детоксикационные ферменты семейства Р450, активность и количество которых значительно возрастает при поступлении в клетку токсических соединений. В настоящее время известно около 150 изоформ Р450, каждая из которых имеет много субстратов для обезвреживания (эндогенные липофильные вещества, лекарственные препараты, этанол, ацетон и др.).
Устранение или уменьшение нарушений обмена воды и электролитов в клетке. В этом принимают участие ряд процессов и механизмов:
· улучшается (активируется) энергообеспечение ионных насосов: Nа+, К+-АТФазы, Са2+-АТФазы. Таким образом, нормализуется содержание ионов Nа, К, Са в клетке. Удаление из клетки Nа+ препятствует излишнему накоплению в ней воды (Н2О уходит за Nа+). Улучшается циркуляция внутриклеточной жидкости, нормализуется объем внутриклеточных структур и клетки в целом;
· активируются механизмы стабилизации внутриклеточного рН. Повреждение клетки часто сопровождается формированием внутриклеточного ацидоза (рН↓). Закисление цитозоля активирует карбонатные, фосфатные и белковые буферные системы клетки. Усиливается работа натрий-водородного противопереносчика (белок NНЕ, Nа+-Н+-обмена), за счет его Н+, в обмен на Nа+ удаляется из цитоплазмы. Активация Nа+-Cl--НСО-3-обменика и Nа+ - НСО-3- котранспортера в клетке увеличивает мощность карбонатного буфера. Повышается уровень гистидиновых дипептидов (карнозина, анзерина, офидина), что значительно усиливает возможности белкового буфера. Например, они создают до 40% буферной емкости быстрых мышц. Кроме того, карнозин активирует работу ионных насосов, стимулирует АТФ-азную активность миозина.
Компенсация расстройств механизмов регуляции внутриклеточных процессов, в том числе и их первичных нарушений (информационной составляющей гомеостаза). Адаптация к данным нарушениям реализуется посредством:
· изменения количества мембранных рецепторов к сигнальным молекулам. В зависимости от ситуации (избыток или недостаток первичных мессенджеров) на поверхности клетки соответственно может уменьшаться или увеличиватся их количество;
· изменения чувствительности мембранных рецепторов к сигнальным молекулам. Изменение количественных и качественных характеристик клеточных рецепторов используется как защитный механизм, например при эндокринопатиях: при гиперпродукции гормонов их количество и чувствительность снижается, а при гипопродукции – увеличивается;

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
