


Тема 6 Исследование биоэквивалентности генерических лекарственных средств.
Большая часть продаваемых в нашей стране лекарственных средств как отечественного, так и импортного производства являются не оригинальными препаратами, а их воспроизведенными аналогами - «генериками». Некоторые фирмы воспроизводят лекарственные средства по лицензии под надзором фирм-разработчиков с полным соблюдением регламента производства. Стоимость выпущенных по лицензии ЛС незначительно отличается от стоимости оригинальных препаратов.
Чаще всего производители выпускают лекарственные средства без покупки дорогостоящей лицензии у патентообладателя. Такие ЛС выпускаются в той же лекарственной форме, содержат такую же дозировку действующих веществ, близкие соотношения вспомогательных веществ, но при этом могут обладать сниженной терапевтической эффективностью. Данный аспект может объясняться несколькими факторами:
Ø Использованием в производстве фармацевтических субстанций с более низкой исходной терапевтической активностью (например, левовращающие оптические изомеры обычно фармакологически более активны; на всасывание в ЖКТ влияет степень измельчения фармацевтических субстанций). При этом и чистота и количественное содержание активного ингредиента в субстанции такое же, как и в спецификации на субстанцию, используемую в производстве оригинального ЛС.
Ø Качеством химических реактивов и вспомогательных веществ, используемых в производстве (наполнители, полимерные пленки, сахара, консерванты, стабилизаторы. Перечисленные вещества могут оказывать серьезное влияние на высвобождение лекарственного вещества из лекарственной формы в организме человека.
Ø Недостаточно высоким обеспечением стабильности технологического процесса производства (недопустимые отклонения (изменения) условий опудривания, прессования, увлажнения и др.).
Согласно Государственной фармакопеи Республики Беларусь «…генерические лекарственные средства должны удовлетворять тем же стандартам качества, эффективности и безопасности, что и оригинальные лекарственные средства, но при этом дополнительно должно быть предоставлено убедительное подтверждение того, что они эквивалентны ранее зарегистрированным аналогичным лекарственным средствам и клинически взаимозаменяемы с ними».
Оценка биоэквивалентности генерических лекарственных средств считается основным видом медико-биологического контроля их эффективности. Оценка биодоступности генерических лекарственных средств выполняется в случаях, если средство содержит одно и то же лекарственное вещество в одинаковой дозе, но при этом имеет отличную лекарственную форму от оригинального лекарственного средства.
Выполнение исследований биоэквивалентности и биодостуности позволяют в гораздо более сжатые сроки и с меньшими материальными затратами сделать обоснованный вывод об их эффективности, чем проведение клинических испытаний.
Биоэквивалентные исследования для генерических лекарственных средств, произведенных разными предприятиями, которые предполагаются для клинической замены, реализуемых на фармацевтическом рынке. Биоэквивалентные исследования назначаются для следующих видов ЛС:
· Любых лекарственных форм, предназначенных для перорального приема, содержащие лекарственные средства с системным действием (таблетки, капсулы, драже, порошки, сиропы, суспензии, эмульсии, микстуры, эликсиры и проч.).
· Непероральных и непарентеральных ЛС, предназначенных для воздействия посредством системной абсорбции (например, мази, гели, биодеградирующие таблетки, суппозитории).
· Медленно высвобождающихся ЛС и других форм препаратов с модифицированным высвобождением активного вещества, которые предназначены для воздействия посредством системной абсорбции (включая транс-дермальные терапевтические системы).
· Фиксированных комбинаций в комплексных средствах системного действия.
· При любых изменениях качественного или количественного состава лекарственного средства (в том числе при изменении соотношения вспомогательных ингредиентов соотношения активных и вспомогательных ингредиентов), выполненные после проведения предшествующих исследований эквивалентности или регистрации лекарственного средства.
Исследования не выполняются для вакцин, сывороток животных, препаратов человеческой крови и плазмы, и препаратов, произведенные при помощи биотехнологии, а также парентеральных и ингаляционных ЛС. Для нерастворимых лекарственных средств, предназначенных для местного (несистемного) действия (мази, пластыри, глазные капли и др.) проводятся сравнительные клинические или фармакодинамические исследования.
При проведении биоэквивалентных исследований используется следующая терминология:
Биологическая доступность (биодоступность, bioavailability) – скорость и степень доступности действующего вещества или его активного компонента (или нескольких) из дозированной лекарственной формы в месте действия, которые определяют с помощью кривой зависимости «концентрация-время» в системном кровообращении или по выделению в моче. При этом считают, что вещество a priori в системном кровотоке находится в динамическом равновесии с веществом в месте действия.
Абсолютную биодоступность данной лекарственной формы определяют путем сравнения с биодоступностью (100%) этого лекарственного средства при условии его внутрисосудистого введения (например, раствор для орального применения в сравнении с раствором для внутривенного введения).
Относительную биодоступность данной лекарственной формы определяют путем сравнения с биодоступностью другой лекарственной формы, введенной тем же или другим (но не внутривенным) путем (например, таблетки в сравнении с раствором для орального применения).
Основой проведения исследований биоэквивалентности и биодоступности генерических лекарственных средств является определение относительной биодоступности.
Биологическая эквивалентность (биоэквивалентность, bioequivalence) – два лекарственных средства биологически эквивалентны, если они фармацевтически эквивалентны или они фармацевтически альтернативны и их биологические доступности (скорость и степень доступности), после приема в одной и той же молярной дозе, похожи до такой степени, что можно предполагать, что их терапевтические эффекты и показатели безопасности будут по существу одинаковыми.
Взаимозаменяемое лекарственное средство – это лекарственное средство, которое терапевтически эквивалентно эталонному лекарственному средству.
Генерическое лекарственное средство (родовой препарат) – это лекарственное средство, содержащее одно и то же лекарственное вещество в одинаковой дозе и в той же лекарственной форме, что и оригинальное лекарственное средство, являющееся биоэквивалентным с оригинальным, производящееся без лицензии от компании, владеющей оригинальным лекарственным средством и продающееся после истечения срока патента или других эксклюзивных прав на оригинальный препарат. Они могут продаваться под международными наименованиями или под новыми торговыми наименованиями.
Допускается присутствие на фармацевтическом рынке генерических средств в дозированных формах и/или количестве отличном от оригинальных лекарственных средств.
Лекарственная форма – это придаваемый лекарственному средству вид, определяющий его состояние, дозировку, упаковку и способ применения.
Оригинальное (исходное, «инновационное») лекарственное средство – это лекарственное средство, которое отличается от всех ранее зарегистрированных лекарственных средств фармакологически активным действующим веществом или комбинацией таких веществ.
Фармацевтически альтернативные лекарства (pharmaceutical alternatives). Лекарственные средства являются фармацевтически альтернативными, если они содержат один и тот же активный компонент (или компоненты), но различаются его химической формой (соль, эфир и др.), лекарственной формой или силой действия (активностью).
Эталонное (справочное) лекарственное средство – это лекарственное средство, с которым предполагается взаимозаменяемость в клинической практике. Эталонное (справочное) лекарственное средство – это обычно оригинальное лекарственное средство, для которого уже установлены эффективность, безопасность и качество. Там, где оригинальное лекарственное средство неизвестно, в его роли может выступать препарат, который лидирует на рынке сбыта, зарегистрирован и разрешен к медицинскому применению с установленной и задокументированной эффективностью, безопасностью и качеством.
Дозировка лекарственного средства – это количество (г, мг, мкг) лекарственного средства в единице лекарственной формы (таблетка, капсула, драже и т. д.).
Доза лекарственного средства – это количество (г, мг, мкг) лекарственного средства на один прием (однократное или многократное применение).
Для фармацевтически эквивалентных или альтернативных лекарственных средств должна быть доказана терапевтическая эквивалентность к другому средству для установления факта взаимозаменяемости. Некоторые методы таких исследований приведены ниже:
· Сравнительные исследования по оценке биологической доступности (биологической эквивалентности) на людях, при которых измеряется концентрация активного ингредиента лекарственного средства либо одного или более метаболитов в доступной биологической жидкости, например, плазма, кровь или моча.
· Сравнительные фармакодинамические исследования на людях.
· Сравнительные клинические испытания.
· Определение сравнительной кинетики растворения in vitro в трех буферных растворах.
Другие методы, которые также могут быть использованы для оценки биологической эквивалентности, например, исследования биологической эквивалентности на животных, допускаются лишь в особо оговоренных случаях.
Выбор процедуры исследования зависит от многих факторов, включая характеристику активного действующего вещества, лекарственного средства и наличие необходимых ресурсов для его проведения.
Исследования сравнительной кинетики растворения (исследования вне живого организма)
Исследования теста сравнительной кинетики растворения выполняется для всех заявляемых на регистрацию дозировок средств в твердых лекарственных формах как этап, предшествующий проведению исследований биологической эквивалентности или биодоступности. Исследование выполняется в трех буферных растворах объемом 250 мл в интервале рН 1-8, при температуре 37°С (предпочтительно при рН около 1,0; 4,6 и 6,8).
Под степенью растворения твердой дозированной формы понимают количество действующего вещества, в процентах, от содержания, указанного в разделе «Состав», которое в условиях, описанных в частной статье, должно перейти в раствор. Обычно для проведения испытания используется прибор с лопастью-мешалкой или корзинкой. Выбор используемого прибора зависит от физико-химических характеристик дозированной формы. Все части прибора, которые могут вступать в контакт с лекарственным средством или средой растворения, должны быть химически инертны, не адсорбировать, не реагировать или каким-либо другим способом искажать результаты испытания. Все металлические части прибора, которые могут вступать в контакт с лекарственным средством или средой растворения, должны быть изготовлены из нержавеющей стали или покрыты соответствующим материалом для того, чтобы эти части не взаимодействовали или каким-либо другим способом не искажали результаты испытания. Прибор должен быть сконструирован таким образом, чтобы свести к минимуму любые колебания и вибрацию, обусловленную проточной системой или элементом, который плавно вращается.
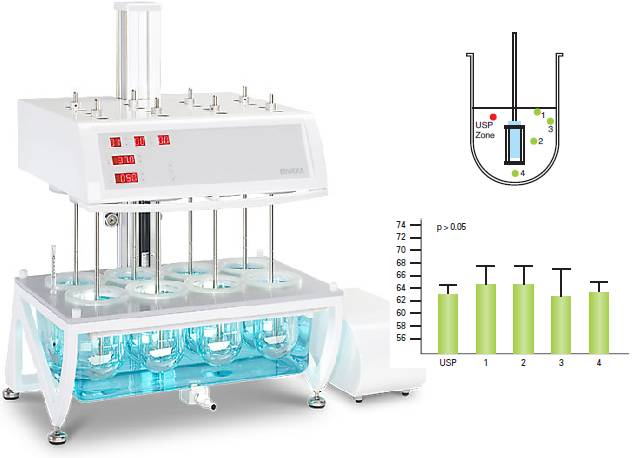
Рисунок – Прибор для проведения теста «Растворение» ERWEKA DT 720 – слева, справа на рисунке изображен сосуд и отмечены пять точек отбора проб, наилучшая воспроизводимость результатов достигается в точке «USP zone».
Прибор для проведения теста «Растворение» состоит из:
· цилиндрического сосуда из боросиликатного стекла или другого подходящего прозрачного материала с полусферическим дном и номинальным объемом 1000 мл; крышки, замедляющей испарение; в крышке должно быть центральное отверстие для оси мешалки и другие отверстия для термометра и устройств, используемых для отбора жидкости;
· водяной бани, которая поддерживает постоянную температуру среды растворения 37,0±0,5°С
· лопасти-мешалки, состоящей из вертикального вала, к концу которого прикреплена лопасть, имеющая форму части круга, отрезанного двумя параллельными хордами;
· мешалки, состоящей из вертикального вала, к нижней части которого прикреплена цилиндрическая корзинка;
· верхняя часть вала должна подсоединяться к мотору, снабженному регулятором скорости; мешалка должна вращаться плавно, без заметных качаний.
Для проведения испытания собирают прибор, нагревают среду растворения до 37±0,5°С и удаляют термометр. Помещают одну единицу испытуемого лекарственного средства в прибор. Следует принять меры, обеспечивающие отсутствие пузырьков воздуха на поверхности лекарственного средства. Вращение лопасти-мешалки или корзинки с указанной скоростью (±4 %) начинают немедленно. Скорость вращения обычно составляет 50 об/мин для лопасти в случае использования прибора с лопастью-мешалкой и 100 об/мин – для корзинки.
Клинический этап исследований
Исследования биоэквивалентности проводятся с одной дозировкой (желательно наибольшей) данного генерического средства, принимаемой натощак, даже если для регистрации он заявлен в нескольких дозировках, только в том случае, если средство находится в лекарственной форме немодифицированного (немедленного) высвобождения; все дозировки данного средства произведены в одинаковых промышленных условиях, по идентичной технологии; соотношение между активным фармакологическим ингредиентом и наполнителями во всех дозировках постоянно (в случае, если количество активного фармакологического ингредиента составляет менее 5% лекарственной формы – соотношение между ингредиентами наполнителей во всех дозировках постоянно); в диапазоне заявленных на регистрацию дозировок зависимость «доза-эффект» для данного лекарственного средства носит линейный характер; профили исследования кинетики растворения для всех заявленных на регистрацию дозировок лекарственного средства в трех буферных растворах эквивалентны.
Назначение исследуемого (тестируемого) препарата и препарата сравнения осуществляется в дозах, не превышающих высшие терапевтические, но достаточных для создания оптимальных условий аналитического определения их (или активных метаболитов) концентраций в биологических жидкостях организма с учетом чувствительности метода и возможностей аппаратуры.
В случае, если на регистрацию заявляется лекарственное средство в кишечнорастворимой оболочке, средство, которое следует принимать после еды, либо пища влияет на биодоступность лекарственного средства, следует проводить исследование с двухкратным приемом лекарства – натощак и через 30 минут после приема горячей, высококалорийной и богатой жирами пищи.
В случае исследования лекарственных форм пролонгированного действия биоэквивалентность следует проверять для каждой дозы в отдельности в условиях однократного и многократного приема (достижения стационарного состояния), если кинетика лекарственного средства нелинейна. В случае, если кинетика лекарственного средства линейна, проводят испытания эквивалентности в условиях однократного приема всех дозировок, а с наибольшей дозировкой дополнительно выполняют исследования в условиях многократного приема (достижения стационарного состояния).
Для трансдермальных лекарственных форм проводят испытания с формой, имеющей максимальную дозировку в условиях однократной и многократной аппликации на идентичные области тела.
Дизайн проведения биоэквивалентных исследований.
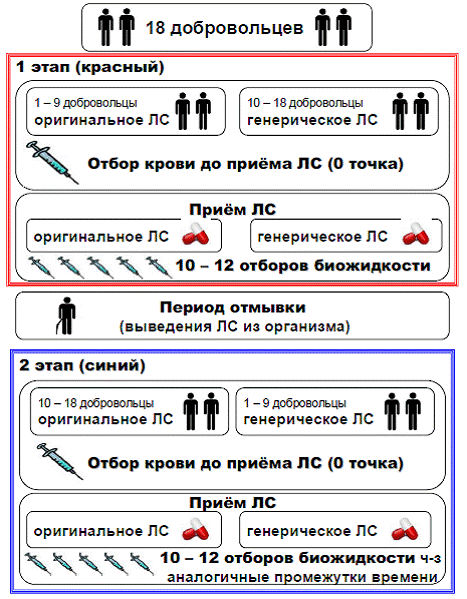
Исследование биологической эквивалентности включает в себя назначение тестируемого и эталонного препаратов в два приема на добровольцах. Промежуток времени между приемом первого и второго лекарственных средств должен составлять не менее шести периодов полувыведения ЛС из организма. Тестируемые лекарственные средства обычно принимаются натощак, запиваются стандартным объемом негазированной воды (150 мл), завтрак может быть получен не ранее чем через четыре часа после начала исследования. За двое суток до начала исследований и в период проведения испытуемые не должны получать другие лекарственные средства, в течение суток до исследования и на всем его протяжении соблюдается строгая диета, исключающая жирную и жареную пищу, напитки, содержащие кофеин, и определенные виды фруктовых соков (например, грейпфрутовый сок). При этом к водному и пищевому режиму предъявляются требования, указанные в протоколе исследования.
Непосредственно перед введением лекарственного средства и через установленные промежутки времени отбираются образцы крови или мочи для определения концентрации лекарственного вещества и метаболитов. При этом график забора крови составляется таким образом, чтобы от «нулевой» точки до максимальной концентрации лекарственного вещества находились, по крайней мере, две точки, еще две точки вблизи максимальной концентрации, а последний забор крови осуществлялся не ранее, чем через четыре периода полувыведения (вся фармакокинетическая кривая после однократного приема ЛС включает не менее 9 точек). Увеличение или уменьшение концентраций лекарственных веществ с течением времени в организме каждого испытуемого позволяет установить кинетику высвобождения лекарственного вещества из тестируемого и эталонного препаратов и охарактеризовать процесс метаболизма. Что, в свою очередь, позволяет провести сравнение двух лекарственных средств и активных метаболитов по кривым зависимости «концентрация-время» в крови или в моче и рассчитать метрические показатели биологической эквивалентности.
Критерии включения добровольцев в исследование.
В качестве здоровых добровольцев могут привлекаться лица обоего пола в возрасте от 18 до 45 лет, отвечающие следующим критериям:
· Верифицированный диагноз «здоров» по данным клинических, лабораторных и инструментальных методов обследования (т. е. не имеющие патологических заболеваний ЖКТ, печени, почек, заболеваний сердечно-сосудистой системы).
· Масса тела не выходит за пределы ±15% по весо-ростовому индексу Кетле.
· Для женщин - отрицательный тест на беременность.
Этические аспекты исследований
Участие здоровых испытуемых в исследованиях биоэквивалентности лекарственных средств является добровольным. Доброволец имеет право на любой стадии отказаться от участия в проводимых исследованиях. Этическую экспертизу клинических исследований биоэквивалентности лекарственных срелств проводит Комитет по этике клинической базы исследований (больница). Добровольцы, включенные в исследование биоэквивалентности, подписывают письменное информированное согласие, один экземпляр которого выдается добровольцу наряду с «Информацией для добровольцев, участвующих в исследовании биоэквивалентности ЛС».
Аналитический этап исследований
Чтобы получить достоверные результаты при проведении биоэквивалентных исследований необходимо использовать биоаналитические методы, должным образом охарактеризованные, полностью валидированные и документированные. Основная цель валидации метода – доказать пригодность выбранного метода для количественного определения концентрации анализируемого вещества (метаболитов) в данном биологическом субстрате. К характеристикам биоаналитического метода, которые важны для обеспечения приемлемости его свойств и достоверности всех результатов анализа, относятся:
· Специфичность.
· Правильность.
· Прецизионность.
· Предел количественного определения и предел обнаружения.
· Линейный диапазон определяемых концентраций лекарственного вещества в биожидкостях.
· Стабильность исследуемых растворов анализируемого вещества в биожидкостях в течение всего периода хранения.
Градуировочный график зависимости аналитического сигнала от концентрации веществ в модельных пробах биожидкостей необходимо строить для каждого анализируемого вещества в каждой серии аналитических определений (не реже одного раза в сутки); его необходимо использовать для вычисления концентрации анализируемого вещества в исследуемых образцах с неизвестным содержанием в данной серии определений. Градуировочный график должен включать «нулевую» концентрацию (blank) и 6-8 образцов с концентрациями анализируемого вещества, охватывающими весь спектр прогнозируемых концентраций в биожидкостях.
Методы пробоподготовки и определения лекарственных веществ и метаболитов
Перечислим кратко основные цели пробоподготовки и требования, предъявляемые к ней.
· вещества, выделяемые в ходе пробоподготовки должны быть пригодны для дальнейшего исследования инструментальными методами, такими как газовая и жидкостная хроматография. То есть, не должны содержать примесей агрессивных веществ, разрушающих хроматографические колонки (таких как молекулярный иод, минеральные кислоты, щелочи, минеральные соли);
· выделение веществ из исследуемых биообъектов должно быть максимально возможным;
· при пробоподготовке должны использоваться доступные и малотоксичные растворители;
· пробоподготовка должна быть по возможности максимально селективной по отношению к определяемым веществам, так чтобы получаемые экстракты содержали минимальное количество сопутствующих веществ, которые будут неминуемо усложнять анализ и интерпретацию результатов;
· в ходе пробоподготовки, по возможности, проводится обогащение пробы определяемыми веществами.
Основные методы:
Прямое осаждение ВМС (хлорной, ТХУ, ТФУ, органическими растворителями). Самый простой по исполнению метод. К анализируемой пробе биожидкости добавляется высаливающий агент, проба встряхивается на миксер-шейкере, центрифугируется, для анализа отбирается надосадочная жидкость. Основной недостаток – необходимость использования высокочувствительного и высокоселективного, а значит и более дорогого аналитического оборудования.

Рисунок – Устройство для перемешивания содержимого пробирок с программируемым таймером и скоростью вращения, а также пробирки с буферными растворами и органическими растворителями для проведения экстракции веществ основного и кислотного характера.
Жидкость-жидкостная экстракция. Достоинством этого вида пробоподготовки можно назвать относительную простоту выполнения, отсутствие необходимости в дополнительном оборудовании (как, например, в случае твердофазной экстракции), использования доступных растворителей, возможность варьировать условия экстракции в зависимости от сложности объекта. Суть метода жидкость-жидкостной экстракции заключается в том, что определяемые вещества, находящиеся в водном растворе переводятся в нейтральную молекулярную форму, путем осторожного подщелачивания среды до рН 9-10 (при определении слабых оснований) или подкисляются до рН 2-3 (при определении слабых кислот), после чего экстрагируется органическим растворителем или смесью, путем перемешивания и центрифугирования. При центрифугировании происходит отделение от минеральной составляющей объекта исследования, которая при этом не переходит в органическую фазу. Органическая фаза отбирается и упаривается, а сухой остаток, содержащий определяемые вещества, растворяется в растворителе для хроматографического анализа.
Твердо-фазная экстракция. Для выполнения необходима камера для твердофазной экстракции, а также картриджи с сорбентом.
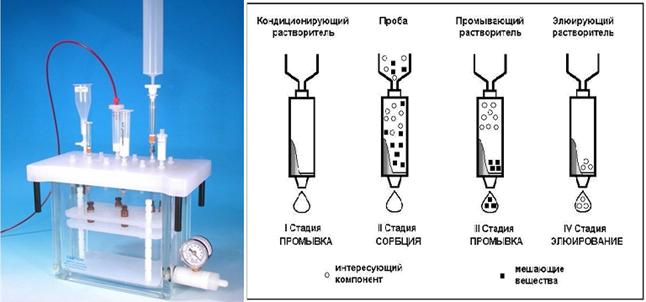
Рисунок – Камера для проведения твердофазной экстракции и четыре последовательных стадии очистки проб
На первом этапе проводится кондиционирование картриджа водно-спиртовой смесью. Эта стадия проводится для активации групп модифицированного силикагеля. На втором этапе через картриджи пропускаются анализируемые образцы, с сорбентом взаимодействуют как определяемые, так и мешающие (балластные) вещества. На третьем этапе происходит промывка картриджа от мешающих определению веществ, используемый при этом растворитель (элюент) подбирается таким образом, чтобы не десорбировать определяемые вещества. На заключительном этапе элюентом с сорбента смываются определяемые вещества.
Методы жидкость-жидкостной и твердофазной экстракции имеется ряд преимуществ и недостатков. К преимуществам первого относится простота выполнения
Основные методы определения лекарственных веществ
· Жидкостная хроматография (со спектрофотометрическим, флуориметрическим, амперометрическим детектированием или с масс-спектрометрическим детектированием (более чувствительный и более дорогой метод – в 10-15 раз);
· Газовая хроматография (пламенно-ионизационное или масс-спектрометрическое детектирование). Основной недостаток анализируемый образец должен быть летучим);
· Капиллярный электрофорез;
· Иммунозимические методы (радиоиммунные и иммуноферментные);
· Биосенсорный анализ (ферментативное оксидазное расщепление глюкозы с образованием пероксида водорода, который и детектируется).
Статистический этап оценки биоэквивалентности
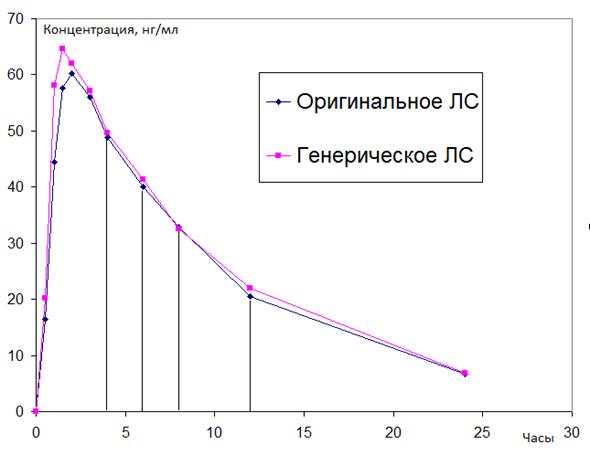
Препараты считаются биоэквивалентными, если: границы оцененного доверительного интервала для Смах, AUC (площади под фармакокинетичекой кривой), как для каждого добровольца, так и для усредненных результатов, находятся в пределах 80-125%;

Все лекарственные средства, разработанные в РБ, перед государственной регистрацией обязательно подвергаются биоэквивалентным или клиническим испытаниям.
